










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkCuadernos de Medicina Forense
versión On-line ISSN 1988-611Xversión impresa ISSN 1135-7606
Cuad. med. forense no.47 Málaga ene. 2007
ATLAS DE PATOLOGÍA FORENSE
Miocardiopatía hipertrófica como causa de muerte súbita en una mujer joven
Hypertrophic cardiomyopathy presenting as sudden death in a young woman
A. Rico1, J. Lucena1, M. Salguero2, M. Blanco1, R. Marín1, E. Barrero1 y MA. Luna1
1 Servicio de Patología Forense. Instituto de Medicina Legal (Sevilla).
2 Servicio de Histopatología. Instituto Nacional de Toxicología y Ciencias Forenses (Sevilla).
Dirección para correspondencia
RESUMEN
La miocardiopatía hipertrófica (MCH) es una enfermedad genética muy heterogénea, con múltiples loci, identificándose para cada gen múltiples mutaciones. Está considerada en Estados Unidos la causa más frecuente de muerte súbita (MS) en jóvenes, fundamentalmente atletas, siendo en ocasiones la primera manifestación de la enfermedad. La MCH se caracteriza morfológicamente por una hipertrofia asimétrica del ventrículo izquierdo y/o derecho con un patrón histopatológico caracterizado por desestructuración (disarray) de los miocardiocitos en una matriz de tejido conectivo prominente así como hipertrofia de la íntima de las arterias coronarias intramurales. Se expone el caso de una mujer joven, de 25 años, diagnosticada clínicamente y con antecedentes familiares de MS por MCH en su madre, que presentó una MS mientras dormía
Palabras clave: Miocardiopatía hipertrófica, muerte súbita, autopsia, patología forense.
ABSTRACT
Hypertrophic cardiomyopathy (HCM) is a genetic and very heterogeneous disease with multiple loci, and with several mutations identified for every gene. In USA, it is regarded as the most common cause of sudden death (SD) in young people (mainly athletes), and occasionally SD is the initial presentation of the disease. HCM is morphologically characterized by an asymmetric hypertrophy of left and/or right ventricle with a histopathological pattern of cardiac myocytes dearrangement in a prominent connective tissue matrix, as well as intimal hypertrophy of intramural coronary arteries. In this paper, we present the case of a 25 year-old woman, clinically diagnosed with HCM, and with family history of HCM in her mother, who experienced a SD while sleeping.
Key words: Hypertrophic cardyomyopathy, sudden death, autopsy, forensic pathology.
Introducción
La Miocardiopatía Hipertrófica (MCH) es una patología heterogénea tanto desde el punto de vista morfológico como desde el punto de vista clínico pudiendo presentarse con sintomatología variada que puede llegar hasta la muerte súbita. La MCH se hereda en un patrón autosómico dominante. En la actualidad se considera que la MCH es una enfermedad del sarcómero ya que todos los genes responsables de la miocardiopatía hipertrófica identificados hasta la fecha codifican proteínas sarcoméricas. Se caracteriza por la hipertrofia del ventrículo izquierdo y/o derecho, usualmente asimétrica y que compromete al tabique interventricular. Histopatológicamente se caracteriza por miocitos hipertróficos que se disponen de forma desorganizada (disarray) en una matriz de tejido conectivo prominente e hipertrofia de la íntima de las arterias coronarias intramurales. La desorganización de los miocitos se considera la característica patológica de la MCH.
Presentación del caso
Mujer de 25 años diagnosticada clínicamente de miocardiopatía hipertrófica obstructiva en tratamiento farmacológico con antiarrítmicos (Amiodarona) y betabloqueantes (Propanolol). La madre había fallecido súbitamente a los 48 años por la misma enfermedad. Una noche salió a cenar con los compañeros de trabajo regresando al domicilio sobre las 4 horas de la madrugada. No manifestó ninguna sintomatología y se acostó en el sofá del salón. Poco después el perro comenzó a ladrar por lo que el padre se levantó encontrándola inconsciente. Se avisó a los servicios de emergencia 061 que se limitaron a constatar el fallecimiento.
Hallazgos de Autopsia
Talla 147 cm, peso 40,6 kg y perímetro de cintura de 69 cm. El IMC era de 18,8.
En el examen externo no se evidenciaron lesiones.
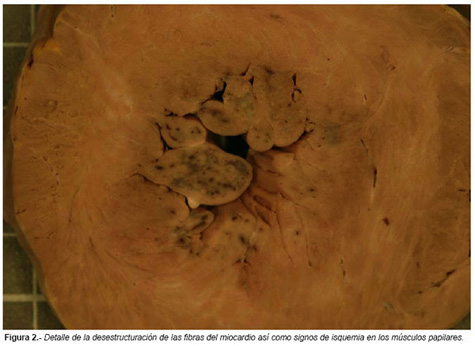
En el examen interno destacaban los hallazgos en el corazón: El corazón pesaba 540 g (peso medio estimado para su peso corporal: 221 g), presentaba hipertrofia del ventrículo izquierdo (Figuras 1 y 2), midiéndose a nivel de la pared lateral un espesor de 2,5 cm y de 2,2 cm en el tabique interventricular. El ventrículo derecho tenía un espesor de 0,6 cm. El tejido miocárdico presentaba zonas de aspecto blanquecino compatibles con fibrosis miocárdica, así como zonas de moteado oscuro, fundamentalmente a nivel de músculos papilares, compatibles con isquemia reciente. La válvula aórtica se encontraba estenosada como consecuencia de la hipertrofia ventricular, así como las valvas de la mitral estaban protruidas con acortamiento de los músculos papilares (Figura 3).


El resultado del análisis toxicológico fue negativo y el estudio histopatológico confirmó el diagnóstico del examen macroscópico, observándose la presencia de miocardiocitos destructurados con ramificaciones anómalas de las miofibrillas (Figura nº 4), así como la presencia de hipertrofia de la media de las pequeñas arteriolas intramiocárdicas (Figura nº 5), típico de la MCH.

Discusión
La Miocardiopatía Hipertrófica (MCH) se caracteriza por la hipertrofia del ventrículo izquierdo y/o derecho, usualmente asimétrica y que compromete al tabique interventricular [1]. Desde el punto de vista histopatológico se caracteriza por miocitos hipertróficos que se disponen de forma desorganizada (disarray) en una matriz de tejido conectivo prominente e hipertrofia de la íntima de las arterias coronarias intramurales. La desorganización de los miocitos se considera la característica patológica de la MCH [1,2]. La MCH es una enfermedad genética cardiaca relativamente común (con una frecuencia de 1:500 en la población general) [3]. Se estima que la prevalencia de MCH diagnosticada mediante ecocardiografía es de 1:1000 en individuos jóvenes y se considera que aumenta con la edad. Este aumento asociado a la edad reflejaría la penetrancia dependiente de la edad de ciertas mutaciones, por ejemplo, las observadas en las mutaciones de la proteína C de unión a la miosina [2]. La muerte súbita es la forma más común de fallecimiento y la complicación más devastadora e impredecible de la MCH [3]. Es el modo de presentación en más del 50% de los pacientes con MCH. Se estima que representa el 5-10% de los casos de muerte súbita en adultos jóvenes, aumentando a un 50% cuando se trata de atletas jóvenes la población estudiada [4]. En el período de octubre de 2003 a diciembre de 2004 supuso el 1% del total de muertes súbitas en la provincia de Sevilla y un 6,25% de los casos en el grupo de ≤ 35 años [5]. En la serie de Basso y cols [6, 7] en ≤ 35 años el porcentaje fue del 5,5%.
La incidencia de muerte súbita entre los pacientes portadores de esta enfermedad varía según las series publicadas. Así, en series que provienen de centros de referencia se encuentran tasas de muerte súbita entre un 3 y un 6% por año. Sin embargo, en poblaciones no seleccionadas la tasa publicada de muerte súbita no superaría el 1% por año. La muerte súbita aparece con más frecuencia en niños y adultos jóvenes entre 14 y 15 años de edad, aunque también puede ocurrir en adultos y es extremadamente rara en niños menores de 5 años, ya que se necesita un período de tiempo suficiente para que la enfermedad desarrolle las alteraciones estructurales que la caracterizan. Habitualmente la muerte súbita es la primera manifestación de la enfermedad, incluso en pacientes asintomáticos previamente diagnosticados. Se han identificado cuatro mecanismos de muerte súbita o síncope en pacientes portadores de MCH: arritmias ventriculares, arritmias supraventriculares causantes de colapso cardíaco, bradicardias e isquemia severa. Entre las características clínicas que marcan peor pronóstico podemos considerar: la edad, per se, representa un primer marcador importante para la misma. De hecho, se registra un 6% de tasa de mortalidad anual en niños y jóvenes frente a un 1% en mayores de 40 años. Los antecedentes familiares de MS son un factor de riesgo clásico para que se produzca dicho evento, demostrándose como el mayor marcador de riesgo de forma individualizada. Así mismo, sobre todo en niños y jóvenes, es importante como factor de riesgo la presencia previa de episodios sincopales. La existencia de taquicardia ventricular sostenida, rara en la MCH, tiene muy mal pronóstico mientras que la taquicardia ventricular no sostenida sólo tiene este mal pronóstico en pacientes con antecedentes sincopales. En cuanto al papel de la isquemia miocárdica como factor de riesgo de la muerte súbita, es dudoso, aunque hay datos que hacen suponer su participación en la misma [8]. Igualmente una disminución de la respuesta vascular periférica es un factor de riesgo para la muerte súbita; pacientes con una anormal respuesta de la presión sanguínea durante el ejercicio tienen un marcado incremento del riesgo a sufrir muerte súbita [9, 10]. Aproximadamente en un tercio de pacientes con MCH falta un incremento de la presión sanguínea apropiado durante el ejercicio. El mecanismo preciso responsable de este control vascular anormal en la MCH es desconocido, si bien es secundario a un aumento de la actividad de los baroreceptores cardíacos [10]. La muerte súbita durante el ejercicio constituye en muchas ocasiones el primer síntoma de la enfermedad [11].
Maron, McKenna et al. [12] en su documento de consenso sobre la MCH establecen una serie de factores de riesgo de MS en la MCH (Tabla nº 1):

La MCH manifiesta una heterogeneidad genética, con múltiples loci, identificándose para cada gen múltiples mutaciones [2]. Al menos se han identificado 10 genes causantes de la enfermedad [13] (Tabla nº 2).

El hecho de que todos los genes asociados con esta patología (beta-MCHC, MBPC, Troponina cardíaca T, Miosina, Actina cardíaca) codifiquen proteínas sarcoméricas define la MCH como una enfermedad del sarcómero. El sarcómero forma la unidad básica de contracción en el músculo cardíaco y, por consiguiente, era razonable postular que el defecto básico es la disfunción contráctil. Los primeros estudios llevados a cabo han puesto de manifiesto que, en realidad, la proteína mutante se incorpora en el sarcómero. Se está examinando la posibilidad de que la proteína mutante afecte al ensamblado de filamentos en el sarcómero. En los casos de MCH humana con mutaciones en beta-MHC, se observa un deterioro en el alineamiento de los filamentos del sarcómero. También aumentó la distancia entre filamentos de miosina próximos y, asimismo, el alineamiento de los filamentos sarcoméricos estaba deteriorado [2].
En el caso que exponemos, se trataba de un cuadro ya diagnosticado, con controles periódicos y tratamiento farmacológico. El fallecimiento se produjo en el propio domicilio de la víctima y en una situación de reposo. Tenía como factores de riesgo el ser una persona joven y fundamentalmente una historia familiar de MS por MCH (el factor de riesgo más importante), así como una obstrucción al flujo de salida del ventrículo izquierdo. El examen necrópsico puso de manifiesto una hipertrofia ventricular asimétrica con afectación de las válvulas aórtica y mitral de carácter obstructivo, compatible con una MCH. El examen histopatológico confirmó el diagnóstico.
El interés médico-forense del caso estribaba en realizar el diagnóstico de una muerte sospechosa fundamentalmente descartando la presencia de tóxicos como causa o factor coadyuvante en el mecanismo de la muerte; y desde el punto de vista sanitario este caso es paradigmático en el sentido de que puede ayudar al clínico a establecer criterios de control y tratamiento en pacientes con una patología susceptible de provocar la MS, ya que sigue siendo uno de los retos fundamentales de la cardiología actual el de construir un algoritmo diagnóstico de la MS que sirva para prevenir este tipo de eventos. q
Bibliografía
1. Report of the 1995 World Hearth Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996; 93:841-842. [ Links ]
2. Roberts R. Genética molecular de las miocardiopatías. Rev Esp Cardiol 2002;55(3):292-302. [ Links ]
3. Maron MJ. Hypertrophic Cardiomyopathy. JAMA 2002;287:1308-1320. [ Links ]
4. Virmani R, Burke AP, Farb A, Atkinson J. Cardiovascular Pathology. Sudden Cardiac Death, Second Edition, vol 40 in the series Major Problems in Pathology. Saunders Company, Philadelphia 2001, pp 340-385. [ Links ]
5. Rico A. Tesis Doctoral: Muerte Súbita del Adulto en la Provincia de Sevilla. Primer Estudio Médico-Forense Realizado en La Comunidad Autónoma Andaluza. Facultad de Medicina; Universidad de Sevilla, 2006. [ Links ]
6. Basso C, Corrado D, Thiene G.Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev 1999 May-Jun;7(3):127-35. [ Links ]
7. Thiene G, Basso C, Corrado D. Cardiovascular Causes of Sudden Death. En: Silver M, Gotlieb A, Schoen T. Cardiovascular Pathology. 3ª ed. Ed. Churchill Livingstone, Philadelphia 2001. Cap. 11. pp 326-74. [ Links ]
8. Gutiérrez Díez A, Tamariz-Martel A, Baño A, Serrano A. Muerte súbita y fibrilación ventricular de posible origen isquémico en un niño con miocardiopatía hipertrófica. Rev Esp Cardiol 2000;53:290-93. [ Links ]
9. Lim PO, Morris-Thurgood JA, Frenneaux MP. Vascular mechanisms of sudden death in hypertrophic cardiomyopathy, including blood pressure responses to exercise. Cardiol Rev 2002 Jean-Feb;10(1):15-23. [ Links ]
10. Elliott P, McKenna WJ. Hypertrophic Cardiommyopathy. Lancet 2004 Jun 5;363(9424):1881-91. [ Links ]
11. Charron P, Komajda M. Hypertrofic Cardiomyopathies. Arch Mal Coeur Vaiss. 2003;96(11):1042-7. [ Links ]
12. Maron BJ, McKenna WJ et al. ACC/ESC Expert Consensus Document on Hypertrophic Cardiomyopathy. JAAC. Vol 42. Nº 9, 2003: 1687-713. [ Links ]
13. Chung MW, Tsoutsman T, Semsarian C. Hypertrophic carddiomyopathy: from gene defect to clinical disease. Cell Research 2003;13(1):9-20. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. Antonio Rico.
Servicio de Patología Forense.
IML de Sevilla.
Avda. Sánchez Pizjuán s/n.
41009 SEVILLA.
Tel. 954370644,
Fax 954906834.
E-mail: iaf.sevilla@andaluciajunta.es

