










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
Print version ISSN 1137-6627
Anales Sis San Navarra vol.26 n.1 Pamplona Jan./Apr. 2003
REVISIONES
Etiopatogenia de la arteritis de células gigantes
Etiopathogenesis of giant cell arteritis
J.M. Casas1, M.V. Acha2
|
RESUMEN La arteritis de células gigantes es una vasculitis que afecta a arterias de diámetro medio y ancho, preferentemente aquellas del arco aórtico con distribución extracraneal, pero también la aorta y otras de sus ramas mayores. Se caracteriza por la presencia de infiltrados inflamatorios mononucleares en la proximidad de la lámina elástica interna constituidos por linfocitos y macrófagos, que en aproximadamente algo más del 50% de los casos contienen células gigantes multinucleadas. La morbilidad asociada a esta enfermedad se relaciona con fenómenos de isquemia distales a la estenosis luminal de las arterias inflamadas y en menor medida con la formación de aneurismas por el debilitamiento de la pared arterial. De etiología desconocida, su patogenia es inmune a través de la migración y localización de células T productoras de γ-INF en la capa adventicia de las arterias inflamadas, suponiéndose que éste es el lugar del estímulo inmune por un antígeno aún no identificado. El reclutamiento y activación de macrófagos por esta citocina constituye uno de los puntos más importantes de su patogenia. La destrucción por éstos del tejido elástico arterial es un fenómeno relevante, así como la producción de otros factores promotores de neoangiogénesis y proliferación de la neoíntima, responsable a través de la obliteración de la luz, de las manifestaciones isquémicas de la enfermedad. El proceso se acompaña de una importante repercusión sistémica caracterizada por una fuerte reacción de fase aguda y síntomas generales de enfermedad poco específicos. Por otra parte, un importante porcentaje de los pacientes presentan un cuadro de polimialgia reumática, entidad en histórica y controvertida relación con esta arteritis. En los últimos años se han producido importantes aportaciones al conocimiento de los mecanismos inmunes implicados en su patogenia. Palabras clave. Arteritis de células gigantes. Patogenesis. Etiología. Epidemiología. |
ABSTRACT Giant cell arteritis is a vasculitis of large and medium size arteries, especially those of the aortic arch with an extracranial distribution, but also the aorta and some of its larger branches. It is characterised by the presence of mononuclear inflammatory infiltrates close to the internal elastic lamina formed by lymphocytes and macrophages, which in slightly more than 50% of the cases contain multinucleate giant cells. The morbidity associated with this disease is related to phenomena of distal ischemia to the luminal stenosis of the inflamed arteries, and to a lesser extent to the formation of aneurisms due to the weakening of the arterial wall. With an unknown aetiology, its pathogenesis is immune through the migration and location of γ-INF -producing T cells in the adventitia of the inflamed arteries, it being assumed that this is the place of immune stimulation by a still unidentified antigen. The recruitment and activation of macrophages by this cytokine is one of the most important points of its pathogenesis. The destruction by these of the arterial elastic tissue is a relevant phenomenon, as is the production of other factors promoting neoangiogenesis and a proliferation of neointime, responsible through obliterating light for the ischemic manifestations of the disease. The process is accompanied by an important systemic repercussion characterised by a strong reaction of acute phase and general but barely specific symptoms of disease. On the other hand, an important percentage of patients show a clinical picture of polymyalgia rheumatica, an entity that has a historical and controversial relationship to this arteritis. In recent years important contributions have been made to the understanding of the immune mechanisms involved in its pathogenesis. Key words. Giant cell arteritis. Pathogenesis. Aetiology. Epidemiology. |
| 1. Servicio de Medicina Interna. Hospital Virgen del Camino. 2. Departamento de Ciencias de la Salud. Universidad Pública de Navarra. | Correspondencia Juan M. Casas Fernández de Tejerina Departamento de Ciencias de la Salud Universidad Pública de Navarra Avda. de Barañáin, s/n 31008 Pamplona E-mail: manuel.casas@unavarra.es |
INTRODUCCIÓN
Las vasculitis son un grupo heterogéneo de síndromes caracterizados por la inflamación de la pared de los vasos sanguíneos. Sus manifestaciones clínicas dependen del calibre y localización de los vasos lesionados y en cierta medida del tipo de inflamación que en ellos se desarrolla. Así, el espectro clínico puede fluctuar desde cuadros autolimitados de carácter benigno con escasas manifestaciones, frecuentemente dérmicas, hasta severos procesos inflamatorios con repercusión sistémica, complicados frecuentemente con fenómenos isquémicos viscerales, distales al lugar de inflamación vascular.
La arteritis de células gigantes (ACG) fue descrita en 1890 por Jonathan Hutchinson como una forma peculiar de arteritis trombosante del anciano, con ocasional producción de gangrena1. Horton en 1932 describió la histopatología típica de la enfermedad y posteriormente otros aspectos de sus manifestaciones clínicas y posibilidades terapéuticas2.
Las vasculitis de grandes vasos incluyen dos entidades típicas, la arteritis de Takayasu y la ACG, caracterizadas por un cuadro inflamatorio granulomatoso con frecuente presencia de células gigantes, que si bien comparten algunos hallazgos histopatológicos y manifestaciones clínicas, son claramente diferenciables entre sí3. Durante las últimas décadas se han identificado otros cuadros de inflamación vascular granulomatosa con presencia de células gigantes en otras localizaciones, como son la arteritis de células gigantes diseminada, la arteritis granulomatosa del SNC, la arteritis focal de células gigantes, la flebitis cutánea primaria, la enfermedad venoclusiva inflamatoria mesentérica y la flebitis de células gigantes del mesenterio4. Existe una potencial superposición entre algunos de estos cuadros no conociéndose con precisión sus interrelaciones, pero aún en su conjunto son muy infrecuentes.
EPIDEMIOLOGÍA
La incidencia de vasculitis en la población general es pequeña, estimándose en 14 casos por 100.000 habitantes de edad superior a los 20 años5, y su aparición puede enmarcarse como un proceso secundario dentro del cuadro clínico de otras enfermedades, frecuentemente infecciosas o autoinmunes y predominantemente cutáneas involucrando pequeños vasos, o más raramente éste es primario, no asociado a otras enfermedades. Entre estas últimas destaca por su mayor incidencia la ACG6, con aparición característica en edades relativamente avanzadas de la vida, pudiendo considerarse ya actualmente, y más aún en un futuro próximo, la vasculitis primaria más frecuente. Debe tenerse en cuenta que en contraste con otras enfermedades vasculares, responsables de una extensa morbimortalidad en el anciano, la arteritis temporal puede ser tratada con éxito.
Esta enfermedad afecta principalmente a personas de raza caucásica, de procedencia norte europea, con una incidencia y prevalencia aproximada de 17-18 por 100.000 habitantes año y de 225 por 100.000 respectivamente, en mayores de 50 años7,8. En algunas series se aprecia una mayor incidencia en mujeres, que triplica la apreciada en hombres (25/100.000 vs 9,4/100.000)9. En regiones del sur europeo se han descrito tasas de incidencia inferiores, próximas a 10/100.000 habitantes de edad superior a 50 años, y también menores diferencias en función del sexo10. A lo largo de las últimas décadas parece haberse incrementado levemente su incidencia11, aunque estos hallazgos pudieran estar influidos por el mayor conocimiento y diagnóstico de la enfermedad.
Se ha demostrado una prevalencia de lesiones histopatológicas de esta arteritis, sin actividad, en pacientes autopsiados próxima al 1,7%, apreciándose que en el 25% de éstos no se había realizado durante su vida el diagnóstico de la enfermedad, lo que sugiere que esta enfermedad puede ser infradiagnosticada o bien, en algunos casos, pasar relativamente desapercibida12.
La media de edad en el momento de aparición de la enfermedad se sitúa alrededor de los 70 años, con muy escasos pacientes en edades inferiores a los 50 años. La supervivencia no parece afectarse por la aparición de la enfermedad, presentando los pacientes con ACG una esperanza de vida similar a su población control por edad y sexo13-15. Algunos estudios apuntan la posibilidad de una mayor incidencia de la enfermedad en el medio urbano, que parece duplicar la observada en el medio rural16,17. Los factores de riesgo vascular clásicos relacionados con la degeneración ateroesclerosa arterial guardan escasa relación con el desarrollo de la enfermedad, excepto en el caso de las mujeres en las que el hábito tabáquico y los antecedentes de enfermedad arterial parecen incrementar el riesgo de desarrollar ACG18.
FACTORES GENÉTICOS Y AMBIENTALES
En el momento actual no existe ninguna evidencia consistente sobre la etiología de esta enfermedad, que pudiera incluso constituir la expresión sindrómica de diferentes agentes etiológicos. La mayor prevalencia de la enfermedad en individuos de origen caucásico norte-europeo y la existencia de casos con agregación familiar19 han sugerido un componente genéticamente determinado en la susceptibilidad a la enfermedad20. Mediante técnicas de microlinfocitotoxicidad, se ha relacionado la enfermedad con el antígeno de histocompatibilidad de clase II HLA-DR421,22, tanto en heterocigotos como en homocigotos, sin demostrarse mayor riesgo o severidad en estos últimos. Las manifestaciones clínicas de la enfermedad parecen ser similares en pacientes portadores de este antígeno que en aquellos en los que está ausente. El estudio de las variantes alélicas del DR4, mediante técnicas de biología molecular, ha permitido apreciar que ciertos subtipos de HLA-DRB1*04, en particular el alelo HLA-DRB1*040423,24, está presente en aproximadamente el 50% de los pacientes y se asocia con una mayor severidad de sus manifestaciones isquémicas25 y con una posible resistencia al tratamiento con esteroides26, aunque no todos los autores han apreciado dicha asociación27 y ésta pudiera estar influida por otros factores étnicos o geográficos28. Otra variante alélica relacionada, pero con una asociación menos clara, es el HLA-DRB1*0101.
En la segunda porción hipervariable de la cadena polimórfica HLA-DRß se identificó una secuencia de cuatro aminoácidos que se asocia a la enfermedad y se localiza en la región de unión con el antígeno, que es distinguible de una secuencia semejante descrita en la artritis reumatoide en la tercera región hipervariable de la misma molécula29, si bien este hallazgo, origen de interesantes hipótesis patogénicas, no ha sido confirmado.
También se ha investigado la asociación de la ACG con polimorfismos de la región -TNF, en desequilibrio de unión/asociación con genes de la región HLA-DR, apreciándose que las formas de ACG sin PMR se asocian con el alelo TNFa2, de forma independiente a su asociación específica con HLA-DRB1*0401 ó *0101, y la PMR pura se asocia con el alelo TNFb330.
La identificación de casos agregados en tiempo y espacio, sin relación familiar, han sugerido la importancia de factores ambientales no conocidos31. Inicialmente Horton y col32 intentaron, infructuosamente, aislar un microorganismo responsable de la enfermedad a través del cultivo e inoculación en animales de material derivado de arterias con la enfermedad. La enfermedad ha sido también relacionada epidemiológicamente con el contacto con periquitos y otros pájaros33. El desarrollo de la enfermedad en el contexto de la estimulación inmune postvacunal ha sido considerado por diferentes autores desde hace décadas, relacionándose específicamente con la vacunación frente a la gripe34-36 y a la fiebre tifoidea37, así como también con la estimulación producida por antecedentes recientes de enfermedad infecciosa38.
La frecuente existencia de unos pródromos inespecíficos de la enfermedad de carácter pseudoviral, la descripción de una posible variación estacional en su incidencia39 y la agregación de casos40, propició el estudio de la hipótesis etiológica viral, de forma paralela a su estudio más fructífero en otros cuadros vasculíticos. Descartada precozmente su relación con la antigenemia del VHB41, se investigó posteriormente su potencial relación con Chlamydia pneumoniae42, con el virus varicella-zoster y con otros agentes infecciosos. Existe evidencia que vincula C. pneumoniae con la enfermedad43,44, pero otros estudios no han confirmado esta relación45,46. La búsqueda de material genético del virus varicela-zoster mediante inmunofluorescencia y PCR en muestras de pacientes con ACG ha sido negativa47. Mediante PCR se ha relacionado de forma preliminar la presencia de DNA del parvovirus B19 con la histopatología de la enfermedad48 y mediante técnicas de hibridación in situ con el citomegalovirus49, aunque posteriormente no han sido apreciados estos hallazgos50,51.
Otras hipótesis identifican como antígenos potenciales a elementos propios de la pared arterial como la elastina o los miocitos de la capa muscular. La llamativa, pero no exclusiva, fragmentación de la lámina elástica interna en la ACG unida a la dificultad de digestión enzimática de la elastina y al agrupamiento de células gigantes alrededor de esta lámina, en un contexto de inflamación granulomatosa, ha alentado desde hace muchos años la hipótesis de un antígeno ligado a la elastina o a péptidos derivados de su degradación. Hazleman en 1975 describió la estimulación de células mononucleares de sangre periférica con un homogeneizado de arteria humana en pacientes con PMR/ACG52. La elastosis actínica de la piel del anciano y la coincidencia de la mayor exposición de la población a la radiación solar en los últimos 50 años con el relativo crecimiento en este periodo de la prevalencia de esta enfermedad en el anciano, ha generado la teoría actínica, en la que se relaciona a la ACG con la exposición, relativamente accesible a la irradiación solar, de la arteria temporal53; que podría en extremo hasta explicar el frecuente carácter asimétrico de las lesiones arteriales, en relación a su variable proximidad a la piel54. Esta hipótesis vincula el daño actínico con la transformación antigénica de la lámina elástica interna arterial55. En esta línea se ha descrito la inducción de respuesta proliferativa in vitro de células mononucleares de sangre periférica por péptidos, derivados de la digestión por la elastasa leucocitaria de la elastina, en el 92% de pacientes con ACG pero sólo en el 9% de un pool de pacientes no-ACG56.
Las células de la capa media son una diana importante del ataque inflamatorio, apreciándose hinchazón, degeneración de células musculares lisas y la unión de macrófagos a estas células57, sin poderse precisar su carácter de agresión primaria o secundaria dentro del proceso de inflamación vascular.
Ocasionalmente se ha descrito la asociación de depósito arterial de amiloide con la ACG58,59, si bien la causalidad de su asociación parece controvertida60.
DISTRIBUCIÓN ANATÓMICA
La ACG es una panarteritis sistémica de carácter granulomatoso que afecta a arterias elásticas de ancho calibre y arterias musculares de mediano calibre. A pesar de su carácter sistémico no se presentan lesiones en todas las arterias musculares, sino que la enfermedad presenta tropismo por ciertos territorios vasculares, manifestándose predominantemente en las arterias del arco aórtico que irrigan específicamente territorios extra-craneales, especialmente en las ramas de la carótida externa. La afectación de arterias intra-craneales por esta enfermedad es una rareza como ha podido apreciarse basándose en estudios necrópsicos de pacientes fallecidos en el contexto de la enfermedad61. Las arterias mas constante (75-100% de casos) y severamente lesionadas en estos estudios fueron la arteria temporal superficial, las arterias vertebrales, la arteria oftálmica y las arterias ciliares posteriores. En el 38-80% de los casos estudiados apreciaron una afectación menos constante y menos intensa en la arteria carótida externa y sus ramas cervicales, los segmentos petroso y cavernoso de la arteria carótida interna y la porción proximal de la arteria central de la retina. Este comportamiento, que respeta exquisitamente las arterias intra-craneales, se ha relacionado con la menor cantidad de tejido elástico de estas arterias y la ausencia de lámina elástica externa en ellas. No obstante, este hallazgo clásicamente aceptado, contrasta en parte con la frecuente lesión de los pequeños vasos próximos a la arteria temporal, que carecen de lámina elástica externa.
Así como en la enfermedad de Takayasu casi todos los pacientes presentan síntomas por la afectación de las grandes ramas del cayado aórtico, en la ACG únicamente el 10-15% de los pacientes los presentarán62. No obstante, en los últimos años se ha relacionado la ACG con la formación de aneurismas63,64 y con el desarrollo de disección o ruptura aórtica65,66. La inflamación de la aorta y de las ramas vasculares de las extremidades superiores ocurre en un grupo peculiar de pacientes, en los que las arterias temporales pueden no mostrar alteraciones hasta en un 42% de los casos y las manifestaciones isquémicas predominan en las extremidades superiores, pudiendo ser mínimas las manifestaciones arteríticas craneales67. La ACG a nivel de las arterias mesentéricas o de las extremidades inferiores es llamativamente rara y las arterias intra-craneales son típicamente respetadas.
Recientemente nuevas tecnologías diagnósticas han permitido conocer la amplitud de la afectación vascular en esta arteritis. La aplicación de la tomografía computerizada helicoidal al diagnóstico de las arteritis de grandes vasos68, ha permitido identificar la afectación aórtica (aortitis) en aproximadamente el 20-40% de los pacientes con ACG, estando precedida frecuentemente por la afectación de las arterias temporales69.
Apoyándose principalmente en la descripción de series cortas o casos aislados, puede valorarse una utilidad prometedora de la RNM, en el diagnóstico de la afectación de las arterias temporales70,71 y de los tejidos próximos a ellas72,73, así como de la ultrasonografía de color duplex74-77.
Mediante tomografía de emisión de positrones con fludeoxiglucosa F18, en un paciente estudiado en un contexto clínico sugestivo de arteritis temporal típica, se apreció una extensa afectación vascular que involucraba la aorta y las arterias subclavias, carótidas, vertebrales y las ilíacas bilateralmente junto a una captación sugestiva de corresponder al tronco principal de la coronaria izquierda78. Esta técnica ha demostrado también captación aumentada del radiotrazador en el bazo79. Utilizando gammagrafía con 67Ga se ha demostrado una captación específica del isótopo en las regiones temporales durante el periodo activo de la enfermedad, remitiendo esta captación a los seis meses de tratamiento con corticosteroides80.
MANIFESTACIONES CLÍNICAS
Las manifestaciones clínicas que presenta esta enfermedad son amplias y pueden clasificarse en tres grupos: 1.- Síntomas arteríticos secundarios a la inflamación vascular focal e isquemia a nivel craneal (alteraciones visuales, claudicación mandibular, ulceración cutaneomucosa, cefalea, engrosamiento arterial, dolor local, etc.); 2.- Síntomas sistémicos (astenia, anorexia, adelgazamiento y fiebre) secundarios a la producción de citocinas; y finalmente, 3.- Síntomas miálgicos.
Existe una amplia superposición entre la Polimialgia Reumática (PMR) y la ACG conocida desde hace más de 50 años que ha suscitado cierta controversia, considerando algunos a la PMR pura como una forma frustrada de Arteritis temporal81. Ambas enfermedades comparten manifestaciones clínicas, una llamativa reacción de fase aguda y una rápida respuesta al tratamiento con corticoesteroides. Aproximadamente el 30-40% de los pacientes con ACG refieren, entre otros, síntomas de PMR82. La relación inversa, pacientes con ACG dentro del grupo de PMR, es llamativamente variable en las series descritas83,84. Recientemente un estudio prospectivo en PMR observa que un 20% de los pacientes presentan de forma concomitante datos clínicos de ambas enfermedades y hasta en el 8% de las presentaciones de PMR clínicamente puras existen datos ecográficos y/o histopatológicos de ACG subyacente85.
La pérdida de visión es una de las más temidas complicaciones isquémicas de la ACG86 que ocurre en aproximadamente el 10-20% de los pacientes87-89. Con menor frecuencia pueden producirse eventos isquémicos cerebrales, especialmente en el territorio vertebrobasilar90. Las alteraciones visuales transitorias están presentes en aproximadamente el 15% de los pacientes y la pérdida completa de visión en el 13%. La trombocitosis presente en algunos de estos pacientes ha sido considerada como un posible factor de riesgo para el desarrollo de estas complicaciones91 y a la inversa la presencia de trombocitosis en pacientes con neuropatía óptica anterior isquémica sugiere la existencia de ACG92. Por otra parte, la existencia de anemia ha sido asociada con un menor riesgo de manifestaciones clínicas visuales severas93. La afectación visual se relaciona con la isquemia del nervio óptico secundaria a vasculitis de la arteria oftálmica, de las arterias ciliares cortas y/o de la arteria central de la retina94,95. En aquellos pacientes con HLA-DRB1*04 se ha descrito una incidencia de manifestaciones oculares tres veces más frecuente y de mayor severidad, así como mayor incidencia de eventos isquémicos neurológicos96. La isquemia ocular del segmento anterior y posterior del globo ocular es extremadamente infrecuente y puede conducir a la ceguera y atrofia óptica97. La amaurosis fugax constituye un predictor de lesiones oculares más graves, que debe ser tenido en cuenta.
PATOLOGÍA MICROSCÓPICA DE LA LESIÓN
La biopsia de la arteria temporal es la prueba que permite la confirmación histopatológica de la enfermedad, si bien existe una amplia variabilidad entre series en el porcentaje de biopsias arteriales normales (42-61%) en pacientes con datos clínicos y analíticos de la enfermedad, asumiendo la escasa especificidad de algunos de éstos98. No obstante, la presencia de manifestaciones clínicas isquémicas es un factor predictivo de positividad de la biopsia99. La posible negatividad de la biopsia se atribuye al carácter segmentario de la enfermedad, aunque éste debería justificar un menor porcentaje100, y se obvia obteniendo muestras de biopsia temporal con tamaños superiores a los dos centímetros. Es importante conocer que el tratamiento esteroideo no modifica a corto plazo la expectativa de positividad de la biopsia en un paciente con sospecha clínica de ACG101.
La ACG puede definirse como una panarteritis linfomonocitaria, con frecuente formación de granulomas, en los que en aproximadamente un 65% de los casos son apreciables células gigantes102. Aunque la histopatología de esta arteritis es especialmente peculiar y característica, dado que la ACG debuta en edades avanzadas, se han de tener presentes los cambios histopatológicos propios del envejecimiento con los que guarda cierta similitud. Lie estudió la repercusión del envejecimiento en la arteria temporal y describió el patrón senil de arteriosclerosis (grado 3), caracterizado por el engrosamiento de la capa íntima, la atrofia de la capa media y la degeneración de las fibras elásticas, que se manifiesta principalmente en la lesión de la membrana elástica interna con aparición de fragmentación, reduplicación, pérdida tintorial y calcificación. Estos hallazgos pueden confundir el diagnóstico especialmente en las formas residuales de la enfermedad.
Los hallazgos histopatológicos pueden agruparse en tres patrones principales. La ACG típica, descrita inicialmente, consistente en una panarterítis granulomatosa con predominio de células mononucleares y presencia de las células gigantes, localizadas principalmente en los alrededores de la membrana elástica interna, con destrucción de ésta, ensanchamiento de la capa íntima con estenosis de la luz arterial y ocasional trombosis, presencia de neoangiogénesis y raramente infiltración de polimorfonucleares con necrosis fibrinoide. La ACG atípica en la que sin identificarse células gigantes, se aprecia el mismo tipo de infiltrado celular, aunque de localización e intensidad más heterogénea; y por último las formas cicatriciales (healed arteritis) que se caracterizan por fibrosis de la pared arterial, de carácter asimétrico, persistiendo en ocasiones cierto grado de infiltrado celular inflamatorio y de neoangiogénesis, que deben diferenciarse de los cambios de arteriosclerosis presentes en estas edades.
Los hallazgos histopatológicos e inmunohistoquímicos sugieren la presencia de una reacción inmune celular. El infiltrado inflamatorio está compuesto mayoritariamente por células T y macrófagos. Los linfocitos colaboradores CD4+ constituyen el 70-90% de las células T que infiltran la pared103. La lesión inflamatoria se distribuye típicamente de forma parcheada y puede constituir bien infiltrados difusos o formar granulomas, pudiendo alternar focos inflamatorios con otros libres de la enfermedad, lo que puede tener relevancia para el diagnóstico en el 10% de los casos100.
Las células de la estirpe monocito-macrófago (CD68+) constituyen el 15-20% del infiltrado104 y están presentes predominantemente en la región interna de la capa media en la proximidad de la lámina elástica interna, que se encuentra fragmentada; en alguna ocasión puede apreciase fagocitosis de fragmentos de ésta por las células gigantes, siendo ésta la localización más frecuente para su visualización. La fragmentación de la lámina elástica interna es un hallazgo de dudosa validez diagnóstica de forma aislada, por su frecuente presencia en arterias temporales seniles.
El análisis morfométrico de las áreas ocupadas por las diferentes capas arteriales demuestra frecuentemente la existencia de un marcado incremento de la capa íntima a nivel de los segmentos arteriales afectados por la enfermedad105,106, que correlaciona con la atrofia de la capa media arterial y la presencia de síntomas arteríticos isquémicos en los pacientes107, en un patrón arterítico denominado por algunos autores como de predominio intimal108. Los cambios en la morfología del vaso secundario a su remodelación, incremento de la capa íntima e inflamación de la media, también pueden ser apreciados mediante biomicroscopía de ultrasonidos109 y mediante algunos métodos de imagen como el TAC helicoidal, la ecografía y la RNM.
En contraste con otras formas de panarteritis, en la ACG es rara la presencia de necrosis fibrinoide en la pared arterial, no así, de neoangiogenesis, extensión de la red vascular más allá de su normal localización restringida a la adventicia hacia la media e íntima arterial. El nivel de neoangiogénesis se relaciona directamente con la presencia de destrucción de la lámina elástica interna, la existencia de amplias neoíntimas y es proporcional a la extensión en superficie de éstas.
Se complica más el diagnóstico de esta enfermedad cuando se consideran otras posibles formas histopatológicas de su expresión, como es el hallazgo aislado de vasculitis de pequeños vasos rodeando una arteria temporal normal. La interpretación de este hallazgo como ACG ha sido recomendado en algunos casos, con la salvedad, especialmente si se objetiva necrosis fibrinoide, de la posible existencia de vasculitis necrotizantes sistémicas en aquellos que presenten alteraciones viscerales110. Otro patrón histopatológico que puede suscitar mayores dudas diagnósticas es la presencia aislada de infiltración inflamatoria perivascular sin otros hallazgos de arteritis111, entidad poco definida y de dudosa filiación que, por otra parte, no asocia una respuesta relevante al tratamiento esteroideo.
PATOGENIA
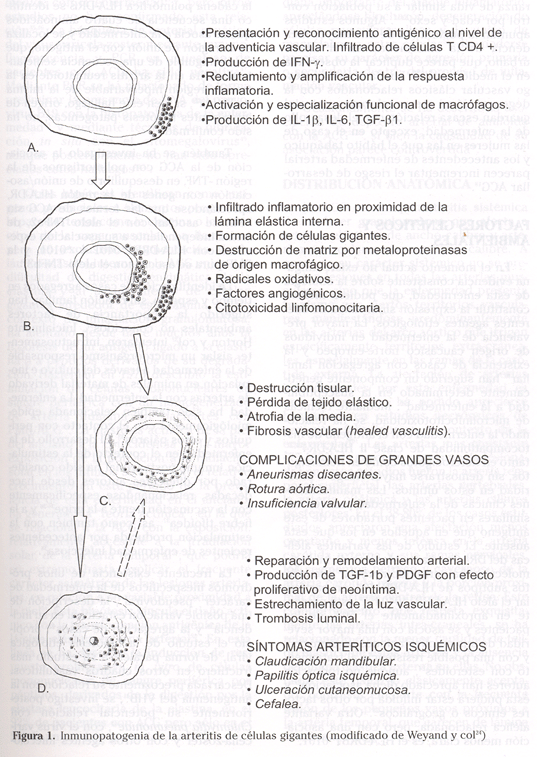
Los conocimientos iniciales sobre la patogenia de algunas vasculitis en los que el depósito de inmunocomplejos en la íntima arterial desencadena el proceso inflamatorio a través de la lesión endotelial, se vieron reforzados por el hallazgo en otras vasculitis de la presencia de anticuerpos específicos frente a las células endoteliales, contribuyendo al modelo patogénico de un agresor centrífugo vehiculado desde el interior del vaso, que tras la lesión endotelial promovía la inflamación vascular. Este modelo es coherente también con la patogenia de la ateroesclerosis, en la que el daño vascular parece generarse desde el interior del vaso. No obstante, en la ACG, contraponiéndose al modelo vasculopático clásico, el inicio de la enfermedad se postula que se localiza en la capa adventicia y la llegada del infiltrado inflamatorio se produce a través de los vasa vasorum.
En la ACG se ha valorado la posible existencia de un fenómeno de reactividad cruzada frente a la pared arterial desencadenada inicialmente en otro lugar de interacción antigénica, pero existen hallazgos que sugieren la presencia del antígeno responsable del fenómeno inflamatorio en la propia pared de la arteria, dado que el transplante de segmentos de ésta en ratones inmunodeficientes no extingue el fenómeno inmune y la arteritis se perpetúa en los injertos transplantados112. Por otra parte, se han observado células reticulares interdigitantes, presentadoras de antígenos, en la pared arterial en un 41% de los pacientes, en mayor número en aquellos con períodos sintomáticos craneales más cortos113, vinculándose este hallazgo al estímulo antigénico en los primeros estadios de la enfermedad. Aunque un 70-90 de los linfocitos que infiltran la pared son CD4+103, con receptor de células T tipo αβ114, la estimulación antigénica se produce a nivel de la adventicia sobre un restringido grupo de linfocitos, aproximadamente un 2-4%, que son CD4+ CD45RO+, que han reorganizado la proteína talina de su citoesqueleto tras reconocimiento antigénico, expresan mayoritariamente CD25+ y un porcentaje próximo al 25% se encuentran en ciclo de división celular (Ki-67+). Estos linfocitos se comportan como los directores de la lesión inmune y desencadenando una respuesta de tipo Th1, con importante secreción de IL-2 y γ-INF, inducen, especialmente por este último, la formación de un proceso inflamatorio de carácter granulomatoso115, con gran activación macrofágica y subsiguiente destrucción y permeabilización de las láminas elásticas y de la capa media arterial por metaloproteinasas.
Algunas de estas células muestran secuencias idénticas en la tercera región determinante complementaria (CDR3) de su receptor clonotípico, aún procediendo de diferentes granulomas o incluso de la arteria temporal contralateral, lo que indica la existencia de proliferación clonal frente a un antígeno presente en la pared arterial116, si bien esta homología del receptor T no es reproducible en diferentes pacientes, lo que sugiere una posible heterogeneidad en el desencadenante antigénico de la reacción inmune, o más probablemente en las características interindividuales de esta reacción.
La migración leucocitaria que integrará el infiltrado inflamatorio se produce predominantemente al nivel de los vasa vasorum de la adventicia en relación con interacciones de las moléculas LFA-1 » ICAM-1 y VLA-4 » VCAM-1, ya que la microvasculatura de la adventicia expresa intensamente moléculas de adhesión endoteliales constitutivas (ICAM1, ICAM2, PECAM y P-selectin) e inducibles (E-selectina y VCAM-1) por el proceso inflamatorio y el infiltrado inflamatorio de la adventicia predominantemente ICAM-3 y las integrinas LFA-1 y VLA-4117.
La producción de γ-INF por las células CD4+ de la adventicia es condición necesaria para el desarrollo completo de la vasculitis, por ser responsable de la inducción del infiltrado inflamatorio y constituir un potente activador macrofágico, considerándosele también el factor fusogénico de macrófagos CD68+ en células gigantes multinucleadas CD68+. Altos niveles de producción local de γ-INF se han asociado con un curso clínico más agresivo y sintomático de la enfermedad, con manifestaciones isquémicas a nivel del territorio de la arteria carótida externa118. La pérdida de la producción in situ de γ-INF previene la aparición de vasculitis y para algunos éste es el factor discriminante respecto de la PMR, a la que consideran forma frustrada de la vasculitis, en la que no se observan infiltrados inflamatorios en su histopatología81,119,120.
Al nivel de la adventicia se produce también un infiltrado de macrófagos (CD68+) productores de Interleucina-1ß (IL-1ß), interleucina-6 (IL-6)104 y factor transformante del crecimiento-1ß (TGF-1ß)121. La producción de IL-6 ha sido demostrada en la pared arterial pero también al nivel de monocitos circulantes activados. Esta citocina además de su papel pro-inflamatorio local está implicada en la relevante producción hepática de reactantes de fase aguda apreciados en la enfermedad. La secreción de IL-1ß y TGF-1ß a nivel de la adventicia puede jugar un papel proinflamatorio a través de la estimulación linfocitaria T y de la quimiotaxis de más células inflamatorias desde los vasa vasorum. El TGF-1ß podría desarrollar a este nivel también funciones de regulación negativa del fenómeno inflamatorio. El papel jugado por la secreción de IL-12 ó IL-10 por los macrófagos, moduladoras positiva y negativa de la secreción de γ-INF por los linfocitos, ha sido estudiado sin poder definir su papel en la variabilidad de expresión clínica de la enfermedad122.
En la capa media, especialmente en la proximidad de la lámina elástica interna y en menor medida de la externa, se produce un importante infiltrado de células de estirpe monocito-macrófago, algunas de las cuales migran hacia esta localización mediante interacción de las moléculas Mac-1≈ ICAM-1 al nivel de la neovascularización que acompaña al proceso inflamatorio en esta localización117. El patrón de actividad de estas células se corresponde únicamente a la secreción de metaloproteinasas, destinadas a la destrucción de la matriz extracelular, especialmente de su componente elástico, permitiendo su permeabilización y la migración del infiltrado inflamatorio y de miofibroblastos a través de la media arterial.
La elastina es el constituyente mayor de las fibras elásticas y se encuentra presente en una alta proporción como proteína estructural de las arterias de gran calibre. Es sintetizada por las células musculares lisas y es crítica para la integridad de muchos tejidos, entre otros de las arterias elásticas. Se trata de una proteína altamente resistente a la proteolisis, únicamente las elastasas de polimorfonucleares y de macrófagos y las metaloproteinasas son capaces de degradarla. Con estas características se han identificado dos metaloproteinasas de la matriz (gelatinasas) de 72 kD y 92 kD, la primera de ellas se expresa constitutivamente por las células del tejido conectivo, músculo liso entre otras, y la segunda es inducible en PMN y macrófagos en respuesta a citokinas liberadas durante la inflamación. Existe un incremento en la transcripción de mRNA de la metaloproteasa-9 de la matriz (colagenasa tipo IV) al nivel de la pared arterial en la ACG. La determinación de esta enzima en el suero de estos pacientes también aprecia valores llamativamente más altos de los observables en sujetos controles123.
Por otra parte, en macrófagos y células gigantes de la media, próximos a la lámina elástica interna, se ha identificado un incremento en la expresión de genes mitocondriales y en la generación de radicales oxidativos, con la subsiguiente aparición en las células próximas, musculares lisas e infiltrado inflamatorio, de lesiones por peroxidación de lípidos124. Se ha apreciado en estas localizaciones la inducción de la enzima aldosa reductasa, cuya función parece estar relacionada con la eliminación de aldehídos tóxicos y productos de peroxidación lipídica, ya que la inhibición experimental de esta enzima se acompaña del incremento de lesiones lipídicas y de células apoptóticas en la pared arterial125.
Las células musculares lisas también participan del fenómeno inflamatorio, ya que la expresión de ICAM-1 en la superficie celular de miocitos próximos al foco de inflamación arterial126, expone a éstos a la citotoxicidad mediada por LFA-1 desde macrófagos o linfocitos T127.
La proteína MCP-1 (monocyte chemoattractant protein) es un importante factor quimiotáctico para células T y macrófagos. Su estudio en la ACG ha apreciado una intensa expresión por las células del endotelio luminal y de los vasa vasorum así como por otras células de la íntima, media y adventicia vascular128. La expresión de MCP-1 indica una participación activa de estas células en el proceso inflamatorio. En otras enfermedades granulomatosas se ha descrito la existencia de relación entre el curso clínico y la concentración plasmática de MCP-1129, que quizá pudiera ser útil en la ACG.
Así, la destrucción celular al nivel de la media está principalmente mediada por macrófagos y células gigantes a través de la producción de metaloproteinasas y radicales libres. En el contexto mencionado la capa media sufre un proceso de atrofia130, verosímilmente responsable del desarrollo de las complicaciones vasculares presentes en esta vasculitis como un fenómeno tardío.
A nivel de la íntima, usualmente aumentada de espesor en mayor o menor medida, se aprecia también frecuentemente un considerable infiltrado de macrófagos cuyo patrón de actividad se dirige a la producción de la sintetasa de oxido nítrico inducible (iNOS). La producción por esta enzima de óxido nítrico, implicado en la regulación del tono vasomotor, se ha descrito también en áreas tisulares dañadas e inflamadas. La producción local de óxido nítrico se ha asociado con efectos pro-inflamatorios y de lesión tisular, a través de la observación de proteínas modificadas por peroxinitritos. No se considera este mecanismo de lesión tisular relevante respecto a la lesión de la capa media, limitándose únicamente a la porción mas interna de la pared arterial131.
En aproximadamente un 50% de los casos al nivel de la lámina elástica interna y de la capa íntima el infiltrado de macrófagos produce TGF-1ß y factor de crecimiento derivado de las plaquetas (PDGF) -AA y -BB132, ambos relacionados con la migración de miofibroblastos desde la capa media arterial y con la aparición de las amplias neoíntimas presentes en algunos de estos pacientes, responsables de la obstrucción luminal y de fenómenos de isquemia característicos de la enfermedad.
Se ha identificado la producción de factor de crecimiento del endotelio vascular (VEGF), por los macrófagos y las células multinucleadas que infiltran la pared arterial en la proximidad de la lámina interna, hecho que contrasta con los hallazgos sobre la angiogénesis en la enfermedad ateroesclerótica donde su producción se debe a células del músculo liso y linfocitos. Por otra parte, la angiogénesis es más prominente en aquellos pacientes que presentan una importante reacción de fase aguda133. Estos hallazgos vinculan fuertemente la neoangiogénesis con el proceso inflamatorio y no meramente con el entorno isquémico acompañante del ensanchamiento de la pared arterial134. La neovascularización se correlaciona estrechamente con la formación de neoíntima, fragmentación de la lámina elástica interna y la presencia de células multinucleares.
Se ha descrito también la existencia de daño directo endotelial, producido por linfocitos activados135 o por un hipotético factor sérico anti-endotelio136, así como la elevación del factor von Willebrand en relación con la fase activa de la enfermedad con normalización de sus valores únicamente tras el establecimiento de remisiones prolongadas137. Hallazgos similares en relación a la implicación de las células endoteliales en la fase activa de la enfermedad, han sido referidos a través del análisis de los niveles de trombomodulina soluble sérica138.
La administración de dexametasona en quimeras de ratones con inmunodeficiencia severa combinada a los que se les injerta un segmento de arteria temporal humana afectada por la enfermedad, demuestra que los esteroides inhiben la síntesis de IL-2, IL-1ß e IL-6 y disminuyen la expresión de la sintetasa inducible de oxido nítrico a través de la inhibición del NFkB, pero apenas inhiben la síntesis de γ-INF y no modifican la expresión de TGF-ß1 en la pared arterial enferma. Con dosis elevadas y prolongadas de esteroides también puede conseguirse, en este modelo, la inhibición de la síntesis de γ-INF pero no de la producción de TGF-ß1 por los macrófagos, a pesar de la disminución de función del infiltrado de células T, ya que si bien el infiltrado de células T persiste a pesar del tratamiento pero su actividad, evaluada por la transcripción de la cadena a del receptor clonotípico, disminuye llamativamente en estas condiciones139. Ulteriores trabajos han demostrado que la producción de γ-INF en este modelo puede ser inhibida por ácido acetilsalicílico a dosis clínicamente relevantes140.
Al nivel de sangre circulante los linfocitos T de los pacientes con ACG muestran marcadores de activación141 como la expresión de HLA-DR, receptor de IL-2, receptor de transferrina y abundante expresión de integrinas142. Algunas de estas células parecen proceder de la activación pauciclonal al nivel de la pared arterial, pudiendo recircular algunas de ellas. Se ha apreciado deplección de la población linfocitaria T CD8+143,144 demostrándose también que la persistencia de un bajo nivel de linfocitos CD8+ en sangre periférica tras seis meses de tratamiento esteroideo, identifica un grupo de pacientes que probablemente experimentarán una enfermedad más severa y precisan de tratamiento más prolongado145.
Al nivel de la circulación el 60-80% de los macrófagos en la ACG y en la PMR se encuentran activados produciendo IL-1ß e IL-6, pero no metaloproteinasas. Esta activación sistémica de los monocitos puede manifestarse de forma independiente a la presencia de vasculitis como sucede en la PMR104 y sugiere dos componentes en la enfermedad, la reacción inflamatoria en la pared vascular y la activación sistémica de los macrófagos.
A escala sistémica se objetiva en sangre periférica la elevación de reactantes de fase aguda (VSG, PCR, haptoglobina, orosomucoide y fibrinógeno) en respuesta a la elevación de IL-6. Los pacientes con una reacción inflamatoria sistémica mas llamativa tienen niveles séricos de IL-6 y TNF-γ más elevados, precisan tratamiento esteroideo más prolongado y presentan más recaídas durante éste146. Durante el tratamiento de la enfermedad las recaídas pueden ser detectadas de forma más sensible por los valores séricos de IL-6 que por la modificación de los reactantes de fase aguda147. También se aprecian niveles elevados del receptor soluble de IL-2 (sIL-2R)148, de ICAM-1149 y de otras moléculas de adhesión. La concentración de sICAM-1 circulante se encuentra relacionada con el curso clínico de la enfermedad y con su respuesta al tratamiento150.
No existe ninguna descripción que objetive la participación del sistema inmune humoral en esta enfermedad, dada la ausencia de linfocitos B o depósito de anticuerpos en las lesiones. No obstante, existen novedosas aproximaciones que tratan de desvelar la relevancia de la respuesta inmune B en esta enfermedad151.
Se ha descrito recientemente un modelo murino de vasculitis de grandes arterias elásticas, producida por el herpesvirus γ68, que identifica al INF-γ como un importante regulador de la inflamación vascular, que evita la persistencia y cronicidad de la infección en las células musculares lisas de estas arterias152. Ratones deficientes en el receptor de INFγ (INF-γR) desarrollan un cuadro de arteritis crónica severa con intensos infiltrados neutrofílicos y/o necrosis en la capa media. Por el contrario, los ratones no deficientes únicamente desarrollan lesiones inflamatorias de carácter leve y predominio mononuclear, sólo cuando reciben dosis mayores de virus infectante. En el ratón deficiente para INF-γR su sistema inmune consigue eliminar el virus en localizaciones como los pequeños vasos, bazo, hígado o pulmones, persistiendo la infección en las grandes arterias elásticas, lo que sugiere que esta localización anatómica se comporta como un lugar inmunoprivilegiado, donde el aclaramiento efectivo de la infección puede conseguirse sólo con la actuación del INF-γ. Este efecto protectivo del INF-γ contrasta con su actuación en la ACG, si bien en esta enfermedad no puede ser evaluada la dificultad de erradicación del potencial agente etiológico, ya que se desconoce la naturaleza de éste, e incluso su hipotético carácter infeccioso. Por otra parte la favorable respuesta a inmunosupresores sitúa probablemente a la ACG en el extremo opuesto a este modelo de infección crónica arterial, con el que probablemente mantendrá únicamente similitud en ciertos mecanismos efectores de la respuesta inmunológica.
En los últimos años se ha producido un progresivo y clarificador avance en el conocimiento de los mecanismos inmunes implicados en esta vasculitis granulomatosa y un sinnúmero de desalentadoras descripciones sobre la presencia/ausencia de diversos microorganismos en la pared arterial de estos pacientes. Verosímilmente el mejor conocimiento de los mecanismos que gobiernan su patogenia a escala celular, servirá para identificar nuevas dianas terapéuticas dentro de los mecanismos de reclutamiento y amplificación inflamatoria, que puedan competir en eficacia y menor incidencia de complicaciones con los corticosteroides, terapéutica hoy por hoy efectiva en la gran mayoría de estos enfermos.
BIBLIOGRAFÍA
1. Hutchinson J. Diseases of the arteries. Nº I. On a peculiar form of thrombotic arteritis of the aged which is sometimes productive of gangrene. Arch Surg (Lond) 1989-90; 1: 323-329. [ Links ]
2. Capobianco DJ, Swanson JW. Neurologic contributions of Bayard T. Horton. Mayo Clin Proc 1998; 73: 912-915. [ Links ]
3. Cid MC, Font C, Coll-Vinent B, Grau JM. Large vessel vasculitides. Curr Opin Rheumathol 1998; 10: 18-28. [ Links ]
4. Weidner N. Giant-cell vasculitides. Semin Diagn Pathol 2001; 18: 24-33. [ Links ]
5. González-Gay MA, García-Porrua C. Systemic vasculitis in adults in northwestern Spain, 1988-1997. Clinical and epidemiologic aspects. Medicine (Baltimore) 1999; 78: 292-308. [ Links ]
6. González-Gay MA, García-Porrua C. Epidemiology of the vasculitides. Rheum Dis Clin North Am 2001; 27: 729-749. [ Links ]
7. Hazleman BL. Polymyalgia rheumatica and giant cell arteritis. En: "Rheumatology" Ed. Klippel JH, Dieppe PA. Esond Edition. Mosby. London 1998; 7.21.1-7.21.8. [ Links ]
8. Nordborg E, Bengtsson BA. Epidemiology of biopsy proven giant cell arteritis. J Intern Med 1990; 227:233-236. [ Links ]
9. Beeson PB. Age and sex associations of 40 autoimmune diseases. Am J Med 1994; 96:457-462. [ Links ]
10. González-Gay MA, García-Porrua C, Rivas MJ, Rodríguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis 2001; 60: 367-371. [ Links ]
11. Machado EB, Michet CJ, Ballard DJ, Hunder GG, Beard CM, Chu CP et al. Trends in incidence and clinical presentation of temporal arteritis in Olmsted County, Minnesota 1950-1985. Arthritis Rheum 1988; 31: 745-749. [ Links ]
12. Ostberg G. On arteritis with special reference to polymyalgia arteritica. Acta Path Microb Scand. 1973; 237 (Suppl.): 1-59. [ Links ]
13. Matteson EL, Gold KN, Bloch DA, Hunder GG. Long-term survival of patients with giant cell arteritis in the American College of Rheumatology giant cell arteritis classification criteria cohort. Am J Med 1996; 100: 193-196. [ Links ]
14. Gran JT, Myklebust G, Wilsgaard T, Jacobsen BK. Survival in polymyalgia rheumatica and temporal arteritis: a study of 398 cases and matched populations controls. Rheumathology (Oxford) 2001; 40: 1238-1242. [ Links ]
15. González-Gay MA, Blanco R, Abraira V, García-Porrua C, Ibáñez D, García-Pais MJ et al. Giant cell arteritis in Lugo, Spain, is associated with low longterm mortality. J Rheumatol 1997; 24: 2171-2176. [ Links ]
16. Reinhold-Keller E, Zeider A, Gutfleisch J, Peter HH, Raspe HH, Gross WL. Giant cell arteritis is more prevalent in urban than rural populations: result of an epidemiological study of primary systemic vasculitides in Germany. Rheumathology 2000; 39: 1396-1402. [ Links ]
17. Schmidt D, Schulte-Mönting J. Giant cell arteritis is more prevalent in urban than in rural populations. Rheumathology (Oxford) 2001; 40: 1193. [ Links ]
18. Duhaut P, Pinede L, Demolombe-Rague S, Loire R, Seydoux D, Ninet J et al. Giant cell arteritis and cardiovascular risk factors: a multicenter, prospective case-control study. Groupe de Recherche sur l'Arterite a Cellules Geantes. Arthritis Rheum 1998; 41: 1960-1965. [ Links ]
19. Liang M, Simkin PA, Hunder GG, Wilske KR, Healey LA. Familial aggregation of polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum 1974; 17: 19-24. [ Links ]
20. González-Gay MA. Genetic epidemiology. Giant cell arteritis and polymyalgia rheumatica. Arthritis Res 2001; 3: 154-157. [ Links ]
21. Cid MC, Ercilla G, Vilaseca J, Sanmarti R, Villalta J, Ingelmo M et al. Polymyalgia rheumatica: a syndrome associated with HLA-DR4 antigen. Arthritis Rheum 1988; 31: 678-682. [ Links ]
22. Sakhas LI, Loqueman N, Panayi GS, Myles AB, Welsh KS. Immunogenetics of polymialgia rheumatica. Br J Rheumatol 1990; 29: 331-334. [ Links ]
23. Weyand CM, Hunder NH, Hicok K, Hunder G, Goronzy J. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis and rheumatoid arthritis. Arthritis Rheum 1994; 37: 514-520. [ Links ]
24. Weyand CM, Hicok HC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. J Clin Invest 1992; 90: 2355-2361. [ Links ]
25. Dababneh A, González-Gay MA, García-Porrua C, Hajeer A, Thomson W, Ollier W. Giant cell arteritis and polymyalgia rheumatica can be differentiated by distinct patterns of HLA clas II association. J Rheumatol 1998; 25: 2140-2145. [ Links ]
26. Rauzy O, Fort M, Nourhashemi F, Alric L, Jucher H, Ecoiffier M et al. Relation between HLA-DRB1 alleles and corticosteroid resistence in giant cell arteritis. Ann Rheum Dis 1998; 57: 380-382. [ Links ]
27. Salvarani C, Boiardi L, Mantovani V, Ranzi A, Cantini F, Olivieri I et al. HLA-DRB1, DQA1, and DQB1 alleles associated with giant gell arteritis in northern Italy. J Rheumatol 1999; 26: 2395-2399. [ Links ]
28. González-Gay MA, García-Porrua C, Hajeer AH, Dababneh A, Ollier W. HAL-DRB1*04 may be marker of severity in giant cell arteritis. Ann Rheum Dis 2000; 59: 574-575. [ Links ]
29. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis: mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest 1992, 90: 2355-2361. [ Links ]
30. Mattey DL, Hajeer AH, Dababneh A, Thomson W, González-Gay MA, García-Porrua C et al. Associationof giant cell arteritis and polymyalgia rheumatica with different tumor necrosis factor microsatellite polymorphisms. Arthritis Rheum 2000; 43: 1749-1755. [ Links ]
31. Rhodes DJ. Giant cell arteritis in general practice. J R Coll Gen Pract 1976; 26: 237-246. [ Links ]
32. Horton BT, Magrath TB. Arteritis of the temporal vessela: report of seven cases. Proc Staff Meet Mayo Clinic 1937; 12: 548-553. [ Links ]
33. Fessel WJ. Polymyalgia rheumatica, temporal arteritis, and contact with birds. Lancet 1969; 2: 1249-1250. [ Links ]
34. Ghose MK, Shensa S, Lerner PI. Arteritis of aged (giant cell arteritis) and fever of unexplained origin. Am J Med 1976; 60: 429-436. [ Links ]
35. Perez C, Loza E, Tinture T. Giant cell arteritis after influenza vaccination. Arch Intern Med 2000; 160: 2677. [ Links ]
36. Finsterer J, Artner C, Kladosek A, Kalchmayr R, Redtenbacher S. Cavernous sinus syndrome due to vaccination-induced giant cell arteritis. Arch Intern Med 2001; 161: 1008-1009. [ Links ]
37. Petersdorf RG, Beeson PB. Fever of unknown origin: report of 100 cases. Medicine 1961; 40: 1-8. [ Links ]
38. Russo MG, Waxman J, Abdoh AA, Serebro LH. Correlation between infection and the onset of the giant cell (temporal) arteritis syndrome. A trigger mechanism? Arthritis Rheum 1995; 38: 374-380. [ Links ]
39. Salvarani C, Gabriel SE, O'Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med 1995; 123: 192-194. [ Links ]
40. Elling P, Olsson AT, Elling H. Synchronous variations of the incidence of temporal arteritis and polymyalgia rheumatica in different regions of Denmark; association with epidemics of Mycoplasma pneumoniae infection. J Rheumatol 1996; 23: 112-119. [ Links ]
41. Bacon PA, Doherty SM, Zuckerman AJ. Hepatitis-B antibody in polymalgia Rheumatica. Lancet 1975; 2: 476-478. [ Links ]
42. Canan Y, Subramaniam S. Chlamydia pneumoniae infection of the central nervous system. Curr Opin Neurol 2001; 14: 355-359. [ Links ]
43. Wagner Ad, Gerard HC, Fresemann T, Schmidt WA, Gromnica-Ihle E, Hudson AP et al. Detection of Chlamydia pneumoniae in giant cell vasculitis and correlation with the topographic arrangement of tissue-infiltrating dendritic cells. Arthritis Rheum 2000; 43: 1543-1551. [ Links ]
44. Rimenti G, Blasi F, Cosentini R, Moling O, Pristera R, Tarsia P et al. Temporal arteritis associated with Chlamydia pneumoniae DNA detected in an artery specimen. J Rheumatol 2000; 27: 2718-2720. [ Links ]
45. Haugeberg G, Bie R, Nordbo SA. Chlamydia pneumoniae not detected in temporal artery biopsies from patients with temporal arteritis. Scand J Rheumatol 2000; 29: 127-128. [ Links ]
46. Regan MJ, Wood BJ, Hsieh YH, Theodore ML, Quinn TC, Hellmann DB et al. Temporal arteritis and Chlamydia pneumoniae: Failure to detect the organism by polymerase chain reaction in ninety cases and ninety controls. Arthritis Rheum 2002; 46: 1056-1060. [ Links ]
47. Nordborg C, Nordborg E, Petursdottir V, LaGuardia J, Mahalingam R, Wellish M et al. Search for varicella zoster virus in giant cell arteritis. Ann Neurol 1998, 44: 413-414. [ Links ]
48. Gabriel SE, Espy M, Erdman DD, Bjornsson J, Smith TF, Hunder GG. The role of parvovirus B19 in the pathogenesis of giant cell arteritis: a preliminary evaluation. Arthritis Rheum 1999; 42: 1255-1258. [ Links ]
49. Fest T, Mougin C, Dupond JL. Giant cell arteritis. Ann Intern Med 1996; 124: 927-928. [ Links ]
50. Helweg-Larsen J, Tarp B, Obel N, Baslund B. No evidence of parvovirus B19, Chlamydia pneumoniae or human herpes virus infection in temporal artery biopsies in patients with giant cell arteritis. Rheumatology (Oxford) 2002; 41: 445-449. [ Links ]
51. Salvarani C, Casali B, Cantini F, Olivieri I, Macchini P, Bajocchi G et al. Detection of parvovirus B19 in temporal arteritis/polymyalgia rheumatica. Arthritis Rheum 2001; 44 Suppl, -S342. [ Links ]
52. Hazleman BL, McLennan ICM, Esiri MM. Lymphocyte proliferation to artery antigen as a positive diagnostic test in polymyalgia rheumatica. Ann Rheum Dis 1975; 34: 122-127. [ Links ]
53. Kinmont PDC, McCallum DI. The aetiology, pathology and course of giant-cell arteritis: the possible role of light sensitivity. Br J Dermatol 1965; 77: 193-202. [ Links ]
54. O'Brien JP, Regan W. A study of elastic tissue and actinic radiation in "aging", temporal arteritis, polymyalgia rheumatica, and atheroesclerosis. The actinic storm in the modern world. J Am Acad Dermatol 1991; 24: 765-776. [ Links ]
55. O'Brien JP, Regan W. Are we losing focus on the internal elastic lamina in giant cell arteritis? Arthritis Rheum 1992; 35: 794-798. [ Links ]
56. Gillot JM, Masy E, Davril M, Hachulla E, Hatron PY, Devulder B et al. Elastase derived elastin peptides: putative autoimmune targets in giant cell arteritis. J Rheumatol 1997; 24: 677-682. [ Links ]
57. Shiki H, Shimokama T, Watanabe T. Temporal arteritis: cell composition and the possible pathogenic role of cell-mediated immunity. Hum Pathol 1989; 20: 1057-1064. [ Links ]
58. Salvarani C, Gabriel SE, Gertz MA, Bjornsson J, Li CY, Hunder GG. Primary systemic amyloidosis presenting as giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum 1994; 37: 1621-1626. [ Links ]
59. Estrada A, Stenzel TT, Burchette JL, Allen NB. Multiple myeloma-associated amyloidosis and giant cell arteritis. Arthritis Rheum 1998; 41: 1312-1317. [ Links ]
60. González-Gay MA, Blanco R, Cereijo MJ, Bal F, González-Vela C. Giant cell arteritis and amyloidosis: comment on the article by Salvarani et al. Arthritis Rheum 1996; 39: 1073-1074. [ Links ]
61. Wilkinson IM, Russell RW. Arteries of the head and neck in giant cell arteritis. A pathological study to show the pattern of arterial involvement. Arch Neurol 1972; 27: 378-391. [ Links ]
62. Klein RG, Hunder GG, Stanson AW, Sheps SJ. Large artery involvement in giant cell (temporal) arteritis. Ann Intern Med 1975, 83: 806-812. [ Links ]
63. Evans JM, O'Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995; 122: 502-507. [ Links ]
64. Nesi G, Anichini C, Pedemonte E, Tozzini S, Calamai G, Montesi GF, Gori F. Giant cell arteritis presenting with annuloaortic ectasia. Chest 2002; 121: 1365-1367. [ Links ]
65. Eklund E, Englund E, Valdemarsson S. Rupture of a non-aneurysmatic aortic trunk in a patient with giant cell arteritis. Ann Rheum Dis 1998; 57: 442-443. [ Links ]
66. Richardson MP, Lever AM, Fink AM, Dixon AK, Hazleman BL. Survival after aortic dissection in giant cell arteritis. Ann Rheum Dis 1996; 55: 332-333. [ Links ]
67. Brack A, Martínez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum 1999; 42: 311-317. [ Links ]
68. Mesurolle B, Qanadli SD, Merad M, El Hajjam M, Mignon F, Lacombe P. Dual-slice helical CT of the thoracic aorta. J Comput Assit Tomogr 2000; 24: 548-556. [ Links ]
69. Evans JM, O'Fallon M, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995; 122: 502-507. [ Links ]
70. Atalay MK, Bluemke DA. Magnetic resonance imaging of large vessel vasculitis. Curr Opin Rheumatol 2001; 13: 41-47. [ Links ]
71. Mitomo T, Funyu T, Takahashi Y, Murakami K, Koyama K, Kamio K. Giant cell arteritis and magnetic resonance angiography. Arthritis Rheum 1998; 41: 1702. [ Links ]
72. Joelson E, Ruthrauff B, Ali F, Lindeman N, Sharp FR. Multifocal dural enhancement associated with temporal arteritis. Arch Neurol 2000; 57: 119-122. [ Links ]
73. Anders HJ, Sigl T, Sander A, Coppenrath E, Pfluger T, Kellner H. Gadolinium contrast magnetic resonance imaging of the temporal artery in giant cell arteritis. J Rheumatol 1999; 26: 2287-2288. [ Links ]
74. Schmidt WA, Kraft HE, Volker L, Vorpahl K, Gromnica-Ihle EJ. Colour Doppler sonography to diagnose temporal arteritis. Lancet 1995; 345: 866. [ Links ]
75. Puechal X, Chauveau M, Menkes CJ. Temporal Doppler-flow studies for suspected giant-cell arteritis. Lancet 1995; 345: 1437-1438. [ Links ]
76. Schmidt WA, Kraft HE, Vorpahl K, Volker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997; 337: 1336-1342. [ Links ]
77. Hunder GG, Weyand CM. Sonography in giant-cell arteritis. N Engl J Med 1997; 337: 1385-1386. [ Links ]
78. Turlakow A, Yeung H, Pui J, Macapinlac H, Liebovitz E, Rusch V, Goy A, Larson S. Fludeoxyglucose positron emission tomography in the diagnosis of giant cell arteritis. Arch Intern Med 2001; 161: 1003-1007. [ Links ]
79. De Winter F, Petrovic M, Van de Wielle C, Vogelaers D, Afschrift M, Dierckx RA. Imaging of giant cell arteritis: Evidence of splenic involvement using FDG positron emission tomography. Clin Nucl Med 2000; 25: 633-634. [ Links ]
80. Généreau T, Lortholary O, Guillevin L, Cacoub P, Galezowski N, Chérin P et al. Temporal 67gallium uptake is increased in temporal arteritis. Rheumathology 1999; 38: 709-713. [ Links ]
81. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med 1994; 121: 484-491. [ Links ]
82. Salvarani C, Hunder GG. Musculoskeletal manifestations in a population-based cohort of patients with giant cell arteritis. Arthritis Rheum 1999; 42: 1259-1266. [ Links ]
83. Salvarani C, Macchioni P, Boiardi L. Polymyalgia rheumatica. Lancet 1997; 350: 43-47. [ Links ]
84. Hunder GG. Giant-cell arteritis and polymyalgia rheumatica. Med Clin North Am 1997; 81: 195-219. [ Links ]
85. Schmidt WA, Gromnica-Ihle E. Incidence of temporal arteritis in patients with polymyalgia rheumatica: a prospective study using colour Doppler ultrasonography of the temporal arteries. Rheumatology (Oxford) 2002; 41: 46-52. [ Links ]
86. Gordon LK, Levin LA. Visual loss in giant cell arteritis. JAMA 1998; 280: 385-386. [ Links ]
87. Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100: 550-555. [ Links ]
88. Liu GT, Glaser JS, Schatz NJ, Smith JL. Visual morbidity in giant cell arteritis. Clinical characteristics and prognosis for visión. Ophthalmology 1994; 101: 1779-1785. [ Links ]
89. Cid MC, Font C, Oristrell J, de la Sierra A, Coll-Vinent B, López-Soto A et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum 1998; 41: 26-32. [ Links ]
90. González-Gay MA, Blanco R, Rodríguez-Valverde V, Martínez-Taboada VM, Delgado-Rodríguez M, Figueroa M et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis. Arthritis Rheum 1998; 41: 1497-1504. [ Links ]
91. Liozon E, Herrmann F, Ly K, Robert PY, Loustaud V, Soria P et al. Risk factors for visual loss in Giant Cell (Temporal) Arteritis: A prospective study of 174 patients. Am J Med 2001; 11: 211-217. [ Links ]
92. Price N, Clearkin LG. Thrombocytosis and giant cell arteritis. Lancet 1994; 343: 672. [ Links ]
93. González-Gay MA, García-Porrua C, Rodríguez-Ledo P, Llorca J. Trends in the inflammatory response in biopsy-proven giant cell arteritis: comment on the article by Cid et al, and the letters by Nesher and Sonnenblick and Liozon et al. Arthritis Rheum 2000; 43: 1427-1428. [ Links ]
94. Kattah JC, Méjico L, Chrousos GA, Zimmerman LE, Manz HJ. Pathologic findings in a steroid-responsive optic nerve infarct in giant-cell arteritis. Neurology 1999; 53: 177-180. [ Links ]
95. Butt Z, Cullen JF, Mutlukan E. Pattern of arterial involvement of the head, neck, and eyes in giant cell arteritis: three case reports. Br J Ophthalmol 1991; 75: 368-371. [ Links ]
96. González-Gay MA, García-Porrua C, Llorca J, Hajeer A, Brañas F, Dababneh A et al. Visual manifestations of giant cell arteritis: trends and clinical spectrum in 161 patients. Medicine (Baltimore) 2000; 79: 283-292. [ Links ]
97. Casson R, Fleming FK, Shaikh A, James B. Bilateral ocular ischemic syndrome secondary to giant cell arteritis. Arch Ophthalmol 2001; 119: 306-307. [ Links ]
98. Duhaut P, Pinede L, Bornet H, Demolombe-Rague S, Dumontet C, Ninet J et al. Biopsy proven and biopsy negative temporal arteritis: differences in clinical spectrum at the onset of the disease. Groupe de Recherche sur l'Arterite a Cellules Geantes. Ann Rheum Dis 1999; 58: 335-341. [ Links ]
99. González-Gay MA, García-Porrua C, Llorca J, González-Louzao C, Rodríguez-Ledo P. Biopsy-negative giant cell arteritis: clinical spectrum and predictive factors for positive temporal artery biopsy. Semin Arthritis Rheum 2001; 30: 249-256. [ Links ]
100. Poller DN, van Wyk Q, Jeffrey MJ. The importance of skip lesions in temporal arteritis. J Clin Pathol 2000; 53: 137-139. [ Links ]
101. Achkar AA, Lie JT, Hunder GG, O'Fallon WM, Gabriel SE. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med 1994; 120: 987-992. [ Links ]
102. Lie JT and Members and Consultants of the American College of Rheumatology Subcommittee on Classification of Vasculitis. Illustrated histopathologic classification criteria for selected vasculitis syndromes. Arthritis Rheum 1990, 33: 1074-1087. [ Links ]
103. Banks PM, Cohen MD, Ginsburg WW, Hunder GG. Immunohistologic and cytochemical studies of temporal arteritis. Arthritis Rheum 1983; 26: 1201-1207. [ Links ]
104. Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest 1994; 94: 1134-1140. [ Links ]
105. Nordborg E, Bengtsson B-A, Nordborg C. Temporal artery morphology and morphometry in giant cell arteritis. APMIS 1991, 99: 1013-1023. [ Links ]
106. Nordborg C, Petursdottir V. Vessel Wall Morphometry in Gian Cell Arteritis. Arthritis Care Res 2000, 13: 286-290. [ Links ]
107. Casas Fernández-Tejerina JM, Acha Arrieta V, García-Bragado Ancín F, Guerrero Setas D. The diagnostic value of arterial wall remodelation in Temporal Arteritis (remitido para publicación). [ Links ]
108. Mambo NC. Temporal (granulomatous) arteritis: a histopathological study of 32 cases. Histopathology 1979; 3: 209-221. [ Links ]
109. Roters S, Szurman P, Engels BF, Brunner R. The suitability of the ultrasound biomicroscope for establishing texture in giant cell arteritis. Br J Ophthalmol 2001, 85: 946-948. [ Links ]
110. Esteban MJ, Font C, Hernández-Rodríguez J, Valls-Solé J, Sanmartí R, Cardellach F et al. Small-Vessel vasculitis surrounding a spared temporal artery: Clinical and pathologic findings in a series of twenty-eigth patients. Arthritis Rheum 2001; 44: 1387-1395. [ Links ]
111. Corcoran GM, Prayson RA, Herzog KM. The significance of perivascular inflammation in the absence of arteritis in temporal artery biopsy specimens. Am J Clin Pathol 2001; 115: 342-347. [ Links ]
112. Brack A, Geisler A, Martínez-Taboada VM, Younge BR, Goronzy JJ, Weyand CM. Giant cell vasculitis as an T cell-dependent disease. Mol Med 1997, 3: 530-543. [ Links ]
113. Cid MC, Campo E, Ercilla G, Palacin A, Vilaseca J, Villalta J et al. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum 1989; 32: 884-893. [ Links ]
114. Schaufelberger C, Stemme S, Andersson R, Hansson GK. T lymphocytes in giant cell arteritic lessions are polyclonal cells expressing ab type antigen receptors and VLA-1 integrin receptors. Clin Exp Immunol 1993; 91: 421-428. [ Links ]
115. Wagner AD, Björnmsson, Bartley GB, Goronzy JJ, Weyand CM. Interferon-g-producing T cells in giant cell vasculitis represent a minority of tissue infiltrating cells and are located distant from the site of pathology. Am J Pathol 1996; 148: 1925-1933. [ Links ]
116. Weyand CM, Schönberger J, Oppitz U, Hunder NNH, Hicok KC, Goronzy JJ. Distinct vascular lessions in giant cell arteritis share identical T cell clonotypes. J Exp Med 1994; 179: 951-960. [ Links ]
117. Cid MC, Cebrián M, Font C, Coll-Vinent B, Hernández-Rodríguez J, Esparza J et al. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte-endothelial cell interactions. Arthritis Rheum 2000; 43: 184-194. [ Links ]
118. Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum 1997, 40: 19-26. [ Links ]
119. Greenbaum LM. Cytokines in polymyalgia and giant cell arteritis. Ann Intern Med 1995; 122: 634. [ Links ]
120. Weyand CM, Goronzy JJ, Hunder GG. Cytokines in polymyalgia and giant cell arteritis. Ann Intern Med 1995; 122: 634. [ Links ]
121. Weyand CM, Wagner AD, Björnsson J, Goronzy JJ. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest 1996; 98: 1642-1649. [ Links ]
122. Weyand CM, Goronzy JJ. Multisystem interactions in the pathogenesis of vasculitis. Curr Opin Rheumatol 1997, 9: 3-11. [ Links ]
123. Sorbi D, French DL, Nuovo GJ, Kew RR, Arbeit LA, Gruber BL. Elevated levels of 92-kd type IV collagenase (matrix metalloproteinase IX) in giant cell arteritis. Arthritis Rheum 1996; 39: 1747-1753. [ Links ]
124. Heike LR, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circulation Res 199; 84: 1050-1058. [ Links ]
125. Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest 1999; 103: 1007-1013. [ Links ]
126. Wawryk SO, Ayberk H, Boyd AW, Rode J. Analysis of adhesion molecules in the immunopathogenesis of giant cell arteritis. J Clin Pathol 1991; 44: 497-501. [ Links ]
127. Makgoba MW, Sanders ME, Ginther Luce GE, Gugel EA, Dustin ML, Springer TA et al. Functional evidence that intercellular adhesión molecule-1 (ICAM-1) is a ligand for LFA-1-dependent adhesion in T cell-mediated cytotoxicity. Eur J Immunol 1988; 18: 637-640. [ Links ]
128. Ellingsen T, Elling P, Olson A, Elling H, Baandrup U, Matsushima K et al. Monocyte chemoattractant protein 1 (MCP-1) in temporal arteritis and polymyalgia rheumatica. Ann Rheum Dis 2000; 59: 775-780. [ Links ]
129. Hashimoto S, Nakayama T, Gon Y, Hata N, Koura T, Maruoka S et al. Correlation of plasma monocyte chemoattractant protein-1 (MCP-1) and monocyte inflammatory protein-1 alpha (MIP-1alpha) levels with disease activity and clinical course of sarcoidosis. Clin Exp Immunol 1998; 111: 604-610. [ Links ]
130. Petursdottir V, Nordborg E, Nordborg C. Atrophy of the aortic media in giant cell arteritis. APMIS 1996; 104: 191-198. [ Links ]
131. Weyand CM, Goronzy JJ. Arterial wall injury in giant cell arteritis. Arthritis Rheum 1999; 42: 844-853. [ Links ]
132. Kaiser M, Weyand CM, Björnsson, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia and ischemic complications in giant cell arteritis. Arthritis Rheum 1998, 41: 623-633. [ Links ]
133. Cid MC, Hernández-Rodríguez J, Esteban MJ, Cebrián M, Gho YS, Font C et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant-cell arteritis. Circulation 2002; 106: 1664-1671. [ Links ]
134. Kaiser M, Younge B, Björnssons, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol 1999, 155: 765-774. [ Links ]
135. Blann AD, Scott DGI. Activated cytotoxic lymphocytes in systemic vasculitis. Rheumatol Int 1991; 11: 69-72. [ Links ]
136. Bocanegra TS, Germain BF, Saba HI. In vitro cytotoxicity to human endotelial cells in polymyalgia rheumatica and giant cell arteritis. Rheumatol Int 1982; 2: 133-136. [ Links ]
137. Cid MC, Monteagudo J, Oristrell J, Vilaseca J, Pallares L, Cervera R et al. Von Willebrand factor in the outcome of temporal arteritis. Ann Rheum Dis 1996; 55: 927-930. [ Links ]
138. Boehme MW, Schmitt WH, Youinou P, Stremmel WR, Gross WL. Clinical relevance of elevated serum thrombomodulin and soluble E-selectin in patients with Wegener's granulomatosis and other systemic vasculitides. Am J Med 1996; 101: 387-394. [ Links ]
139. Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ. Glucocorticoid-mediated represion of citokine gene transcription in human arteritis-SCID chimeras. J Clin Invest 1997; 99: 2842-2850. [ Links ]
140. Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum 2002; 46: 457-466. [ Links ]
141. Cid MC, Campo E, Ercilla G, Palacín A, Vilaseca J, Villalta J et al. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum 1989; 32: 884-893. [ Links ]
142. Wawryk SO, Ayberk H, Boyd AW, Rode J. Analysis of adhesion molecules in the immunopathogenesis of giant cell arteritis. J Clin Pathol 1991; 44: 497-501. [ Links ]
143. Macchioni P, Boiardi L, Salvarani C, Rossi F, Casadei-Maldini M, Mancini R et al. Lymphocyte subpopulations analysis in peripheral blood in polymyalgia rheumatica/giant cell arteritis. Br J Rheumatol 1993; 32: 666-670. [ Links ]
144. Pountain G, Keogan M, Hazleman B, Brown D. Lymphocyte subpopulations analysis in peripheral blood in polymyalgia rheumatica/giant cell arteritis. Br J Rheumatol 1994; 33: 194-195. [ Links ]
145. Salvarani C, Boiardi L, Macchioni P, Rossi F, Tartoni P, Casadei Maldini M et al. Role of peripheral CD8 lymphocytes and soluble IL-2 receptor in predicting the duration of corticosteroid treatment in polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis 1995; 54: 640-644. [ Links ]
146. Hernández-Rodríguez J, García-Martínez A, Casademont J, Filella X, Esteban MJ, López-Soto A et al. A strong initial systemic inflammatory response is associated with higher corticosteroid requirements and longer duration of therapy in patients with giant-cell arteritis. Arthritis Rheum 2002; 47: 29-35. [ Links ]
147. Caplanne D, Le Parc JM, Alexandre JA. Interleukin-6 in clinical relapses of polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis 1996; 55: 403-404. [ Links ]
148. Pountain G, Hazleman B, Cawston TE. Circulating levels of IL-1beta, IL-6 and soluble IL-2 receptor in polymyalgia rheumatica and giant cell arteritis and rheumatoid arthritis. Br J Rheumatol 1998; 37: 797-798. [ Links ]
149. Macchioni P, Boiardi L, Meliconi R, Salvarani C, Grazia Uguccioni M, Rossi F et al. Elevated soluble intercellular adhesion molecule 1 in the serum of patients with polymyalgia rheumatica: influence of steroid treatment. J Rheumatol 1994; 21: 1860-1864. [ Links ]
150. Coll-Vinent B, Vilardell C, Font C, Oristrell J, Hernández-Rodríguez J, Yague J et al. Circulating soluble adhesion molecules in patients with giant cell arteritis. Correlation between soluble intercellular adhesion molecule-1 (sICAM-1) concentrations and disease activity. Ann Rheum Dis 1999; 58: 189-192. [ Links ]
151. Schmits R, Kubuschok B, Schuster S, Preuss KD, Pfreundschuh M. Analysis of the B cell repertoire against autoantigens in patients with giant cell arteritis and polymyalgia rheumatica. Clin Exp Immunol 2002; 127: 379-385. [ Links ]
152. Dal Canto AJ, Swanson PE, O'Guin AK, Speck SH, Virgin HW. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest 2001; 107: 15-22. [ Links ]

