










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versão impressa ISSN 1137-6627
Anales Sis San Navarra vol.28 no.3 Pamplona Set./Dez. 2005
REVISIONES
Linfoma cerebral primario: revisión bibliográfica y experiencia en el Hospital de Navarra
en los últimos 5 años (2000-2004)
Primary CNS Lymphoma: bibliographical review and experience at the Hospital of Navarre
in the last 5 years (2000-2004)
I. Zazpe1, P. de Llano2, A. Gorosquieta3, T. Cabada4, T. Tuñón2, A. Vázquez1, J. Azcona1, A. Gallo-Ruiz1, E. Portillo1
| RESUMEN El linfoma cerebral primario (PCNSL, Primary CNS Lymphoma) es un linfoma no Hodgkin agresivo que se origina en el sistema nervioso central sin evidencia de linfoma en otra localización en el momento del diagnóstico. Los linfomas cerebrales primarios son menos conocidos y caracterizados que sus homólogos los linfomas sistémicos, por tratarse de una entidad poco frecuente hasta hace unas décadas. Sin embargo, el gran ascenso de incidencia que está experimentando esta patología en las últimas tres décadas y que aún no ha sido explicado, hace necesario la existencia de más estudios para conocer mejor la etiopatogenia de esta entidad. Gracias a los nuevos regímenes de tratamiento, el pronóstico de esta patología ha mejorado significativamente en los últimos años. Sin embargo, el tratamiento del linfoma cerebral primario continúa planteando numerosas controversias en la actualidad, debido a su elevada neurotoxicidad en los pacientes mayores de 60 años, grupo de pacientes frecuentemente afectado por esta patología. Para resolver éstas y otras cuestiones se precisa ahondar en el estudio del linfoma cerebral primario y la realización de potentes ensayos clínicos. Palabras clave. Linfoma cerebral primario. Tumores fantasma. Protocolo DeAngelis. | ABSTRACT Primary cerebral lymphoma (Primary CNS Lymphoma, PCNSL) is an aggressive non-Hodgkin lymphoma that originates in the central nervous system without evidence of lymphoma in any other localization at the time of diagnosis. Primary cerebral lymphomas are less well-known and are characterized than their homologues the systemic lymphomas, as they are an entity whose frequency was scarce until a few decades ago. However, the great rise in incidence that this pathology has undergone over the last three decades, and which is still unexplained, makes more studies necessary to better understand the etiopathology of this entity. Thanks to the new systems of treatment, the prognosis of this pathology has improved significantly in recent years. Nonetheless, treatment of primary cerebral lymphoma continues to give rise to numerous controversies at present due to its high neurotoxicity in patients over 60 years of age, a group of patients frequently affected by this pathology. To resolve these and other questions it is necessary to deep in the study of primary cerebral lymphoma and to carry out high quality clinical trials. Key words. Primary Central Nervous System Lymphoma. Ghost tumors. DeAngelis Protocol. |
| 1. Servicio de Neurocirugía. Hospital de Navarra. Pamplona. 2. Servicio de Anatomía Patológica. Hospital de Navarra. Pamplona. 3. Servicio de Hematología. Hospital de Navarra. Pamplona. 4. Servicio de Radiología. Hospital de Navarra. Pamplona. Aceptado para su publicación el 19 de agosto de 2005 | Correspondencia Idoya Zazpe Cenoz Servicio de Neurocirugía Hospital de Navarra Irunlarrea, 3 31008 Pamplona Tfno. 848 42 21 90 E-mail: idoyaz@yahoo.es |
INTRODUCCIÓN
El linfoma cerebral primario (PCNSL, Primary CNS Lymphoma) es un linfoma no Hodgkin agresivo que se origina en el cerebro, ojos, leptomeninges o médula espinal sin evidencia de linfoma en otra localización en el momento del diagnóstico1. Se trata de neoplasias originadas normalmente por células de tipo B y que resultan indistinguibles microscópica e inmunológicamente de los linfomas sistémicos no Hogdkin. En contraposición, los linfomas cerebrales secundarios resultan de la extensión al Sistema Nervioso Central (SNC) de un linfoma sistémico. Determinados tipos histológicos de linfoma sistémico, como el linfoma de Burkitt y el linfoma linfoblástico del manto y linfomas de determinadas localizaciones, como el anillo de Waldeyer, mama, testículos y médula ósea, tienen especial tropismo para extenderse al SNC, siendo estos linfomas secundarios los linfomas más frecuentes a nivel del SNC. Se desconoce por qué el PCNSL se desarrolla primariamente en un órgano que carece de sistema linfático. Se han postulado diversas hipótesis como: que las células tumorales se originen en un órgano linfoide y posteriormente se asienten en el SNC por determinadas moléculas de adhesión, que dichas células sean sistemáticamente erradicadas por un sistema inmune intacto, pero quedasen relativamente protegidas en el interior del SNC, o que un proceso inflamatorio policlonal intracerebral progrese a una expansión monoclonal neoplásica.
Los PCNSL fueron descritos por primera vez por Bailey en 1929 como sarcoma peritelial. Hemos centrado nuestro estudio en los PCNSL por ser menos conocidos y caracterizados que sus homólogos sistémicos, ya que hasta hace tres décadas se trataba de una entidad muy poco frecuente; constituían el 1% de de los tumores cerebrales primarios y el 1% de los linfomas. Sin embargo, el gran ascenso de incidencia que está experimentando esta patología en la actualidad y que aún no ha sido explicado, hace necesario la existencia de más estudios para conocer mejor la etiopatogenia de esta entidad. Su incidencia se ha duplicado en las últimas dos décadas, constituyendo en la actualidad un 2% de los linfomas malignos y un 3% de los tumores primarios del SNC2. Su incidencia aproximada es de 30 casos de PCNSL por millón de habitantes y año y se han convertido en el segundo tumor maligno cerebral más frecuente en USA, sólo por detrás de los gliomas2. Este aumento de la incidencia en las últimas décadas pudo ser debido, en gran parte, a la epidemia de VIH. Sin embargo, en la actualidad parece haber disminuido entre los individuos VIH positivos gracias a los avances en la terapia antirretroviral y se ha triplicado entre los individuos inmunocompetentes mayores de 60 años sin conocerse aún el motivo2,3. Afectan más frecuentemente a los varones con una ratio 1,5:1. El pico de incidencia se sitúa en la sexta década de la vida, siendo de aparición más precoz en pacientes inmunodeprimidos (primera década en inmunodeficiencias congénitas y cuarta década en pacientes transplantados y con SIDA). Si bien los PCNSL pueden afectar tanto a pacientes inmunocompetentes como inmunodeprimidos, determinadas condiciones asociadas a inmunosupresión conllevan un mayor riesgo de padecer un linfoma primario del SNC, como:
– Conectivopatías autoinmunes: Lupus eritematoso sistémico (LES), S. Sjögren, artritis reumatoide (AR).
– Pacientes transplantados, síndromes de inmunodeficiencia congénitos (S. Wiskott-Aldrich) y SIDA. El riesgo de padecer un PCNSL en un individuo transplantado es 100 veces mayor que el de la población inmunocompetente, debido al desarrollo de potentes agentes inmunosupresores para la prevención del rechazo en estos pacientes.
– Respecto al SIDA, la aparición de un PCNSL se da en los estadios de más severa inmunodepresión (CD4 <50/mm3)2.
– Infección por virus de Epstein-Barr4 (presente en las células tumorales en más del 95% de los casos en individuos inmunodeprimidos, frente a menos del 20% en inmunocompetentes).
CLÍNICA
Pueden causar sintomatología clínica similar a cualquier otra lesión ocupante de espacio intracraneal. La forma clínica de debut más frecuente es la focalidad neurológica (50-80%). Otras formas de presentación clínica pueden ser: síntomas neuropsiquiátricos (20-30%), hipertensión intracraneal (10-20%) y crisis epilépticas (5-20%). Pueden debutar con afectación ocular (uveítis o linfoma vítreo), la cual ocurre en alrededor de un 10% de los casos y puede anteceder a la afectación intracraneal5. Debido a la tendencia de estos tumores a la invasión leptomeníngea, pueden producir déficit de múltiples pares craneales por meningitis carcinomatosa y clínica de compresión medular por afectación leptomeníngea a nivel espinal. El tiempo medio entre el comienzo de los síntomas y el diagnóstico es de 2 meses.
NEUROIMAGEN
Los PCNSL suelen presentarse más frecuentemente como lesiones intraparenquimatosas, en contraste con los linfomas secundarios que tienden a presentarse más frecuentemente con invasión leptomeníngea. Los linfomas primarios del SNC son supratentoriales en aproximadamente un 85% de los casos e infratentoriales en 15% de los casos (Fig. 1). Las localizaciones más frecuentes son los lóbulos frontales, los núcleos profundos de la base y a nivel periventricular (Figs. 2 y 3). Entre un 25 y un 50% de los pacientes pueden tener lesiones múltiples en el momento del diagnóstico; esta cifra puede alcanzar el 85% en individuos inmunodeprimidos.
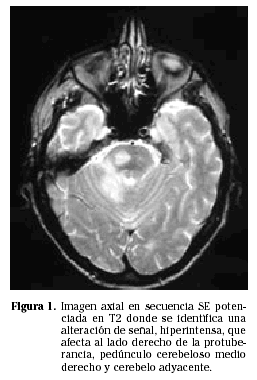
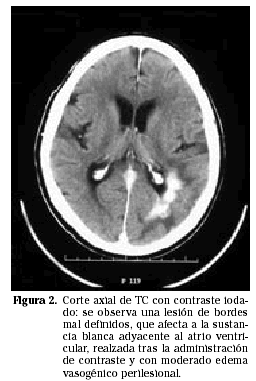
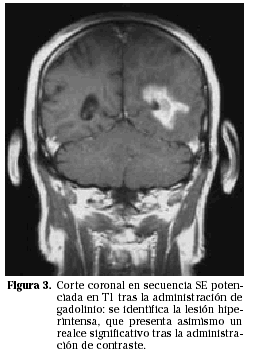
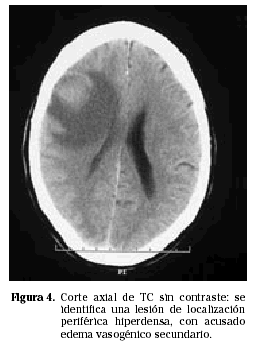
EVALUACIÓN SISTÉMICA
Todos los pacientes con diagnóstico o con sospecha diagnóstica de linfoma deben ser historiados, explorados y sometidos a test de laboratorio (incluida serología del virus de inmunodeficiencia humana¯VIH) para descartar la presencia de inmunosupresión. Asimismo debe descartarse la existencia de un linfoma sistémico, ya que los linfomas primarios del SNC son poco frecuentes y muchos pacientes con linfoma cerebral tienen un linfoma sistémico (linfoma cerebral secundario). El examen incluirá idealmente: examen de nódulos linfáticos, examen de nódulos perihiliares y pélvicos (TC toraco-abdomino-pélvico), analítica de sangre y orina, biopsia de médula ósea coxal y ecografía testicular en varones10.
Además todos los pacientes deben ser sometidos a un examen oftalmológico para descartar infiltración ocular, análisis de LCR (mediante punción lumbar si no existe efecto masa) para valorar posible infiltración leptomeníngea y RMN espinal para descartar infiltración a ese nivel11. Hasta en un tercio de los casos de PCNSL en pacientes inmunocompetentes pueden encontrarse células malignas en líquido cefalorraquídeo (LCR) tras punción lumbar11.
ANATOMÍA PATOLÓGICA
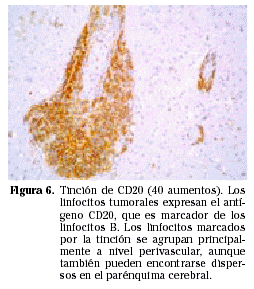
Las células neoplásicas son idénticas a las de los linfomas sistémicos. La mayoría son linfomas no Hodgkin de grado alto o intermedio de malignidad, de distribución perivascular y con marcadores de superficie de linfocitos tipo B12 (Figs. 5 y 6). Excepcionalmente se han encontrado PCNSL originados a partir de células linfoides tipo T13 y casos de linfomas de bajo grado14.
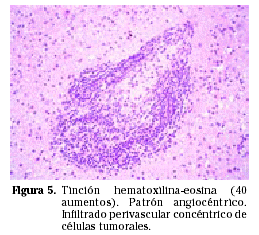
Macroscópicamente se asemejan a los gliomas de alto grado, pueden tener carácter infiltrativo y zonas necróticas.
Microscópicamente están constituidos por masas de células linfoides densamente pobladas, especialmente en el centro del tumor donde incluso llega a desaparecer la estructura del parénquima cerebral. Las células linfoides se disponen típicamente alrededor de los vasos (disposición perivascular) (Fig. 6). Las células se asientan en los espacios de Virchow-Robin, donde separan las fibras de reticulina existentes a la vez que inducen la formación de nuevas fibras, dando lugar a múltiples anillos concéntricos de reticulina en los espacios perivasculares. Esta disposición se hace más evidente en la periferia del tumor, donde la densidad celular es menor. Aunque se trata de masas bien definidas, no es raro encontrar invasión tumoral más allá de los márgenes macroscópicos15. La necrosis coagulativa se da con mayor frecuencia en los casos asociados a inmunodepresión15.
GENÉTICA MOLECULAR
En comparación con otros linfomas originados fuera del SNC, la biología molecular y la genética de los PCNSL no ha sido bien caracterizada16. Se desconoce con precisión el espectro mutacional de los oncogenes y genes supresores en el PCNSL17. Son pocos los estudios moleculares y de citogenética llevados a cabo en PCNSL18. Las pérdidas de material genético más frecuentes se sitúan en el cromosoma 6q18 y las ganancias más frecuentes a nivel de 12q18. Las ganancias cromosómicas son más frecuentes que las pérdidas18. Las pérdidas de material en la región cromosómica 6q se han relacionado con un peor pronóstico18. Asimismo se ha relacionado la metilación del promotor del gen del transportador del folato reducido con resistencia al metotrexate en PCNSL19.
Algunos estudios han mostrado frecuentes anomalías (90%) e inactivación (40-60%) en el gen supresor p14, las cuales son infrecuentes en los linfomas sistémicos, lo cual sugiere un diferente mecanismo de carcinogénesis en los PCNSL y en los linfomas sistémicos20. El mecanismo etiopatogénico de los PCNSL también parece ser distinto en individuos inmunosuprimidos e inmunocompetentes21.
DIAGNÓSTICO Y TRATAMIENTO
El tratamiento se basa en la corticoterapia, la quimioterapia y la radioterapia, puesto que se trata de tumores muy sensibles a estos tres tratamientos. Estos tumores responden bien inicialmente, pero recidivan al tiempo (hasta el 90% durante el primer año). La recidiva suele producirse dentro del SNC, pero, al contrario que en los gliomas, puede producirse a distancia del lugar de aparición inicial.
La biopsia estereotáctica es normalmente adecuada para el diagnóstico de estos tumores normalmente profundos22. Es conveniente realizar una biopsia precoz o retirar los corticoides previamente a la realización de una biopsia (excepto si existe riesgo de herniación cerebral) por la tendencia de estos tumores a producir remisiones con corticoides (ghost tumors)9. La resección quirúrgica parcial o completa de estos tumores no cambia el pronóstico. La cirugía resectiva se realiza normalmente como forma de diagnóstico en caso de lesiones únicas accesibles quirúrgicamente, de las que normalmente no se sospechaba el diagnóstico de linfoma cerebral como primera opción diagnóstica o en casos de urgencia por gran efecto masa con herniación cerebral. La colocación de un sistema de acceso intraventricular con reservorio subcutáneo (Ommaya) para la administración de metotrexate (MTX) intraventricular forma parte del tratamiento de estos tumores y mejora el pronóstico23.
Se ha comprobado mayor supervivencia con tratamiento combinado de radioterapia y quimioterapia que con radioterapia sola y mejor pronóstico si, además de quimioterapia sistémica se emplea quimioterapia con MTX intraventricular mediante reservorio Ommaya y no simplemente intratecal (mediante punción lumbar)23. Gracias a este cambio en la línea de tratamiento la supervivencia media de estos pacientes se ha triplicado en la última década: de 1 año con radioterapia holocraneal a altas dosis a más de 3 años con los nuevos regímenes de tratamiento2, que además intentan minimizar al máximo la neurotoxicidad. Los fármacos quimioterápicos utilizados deben ser capaces de atravesar la barrera hematoencefálica (BHE), como el MTX , la ciclofosfamida y el AraC24. Los protocolos estándar de tratamiento actual de los linfomas cerebrales primarios asocian altas dosis de MTX y radioterapia holocraneal25,26. Se basan, con más o menos variaciones, en el protocolo establecido por DeAngelis27,28 que es el utilizado en nuestro centro y que resumimos a continuación.
Tras la confirmación diagnóstica por biopsia y la colocación de un reservorio Ommaya, se instaura tratamiento con corticoides. Se inicia tratamiento quimioterápico con metotrexate iv (vía eficaz para el tratamiento de la afectación intraparenquimatosa) e intraventricular a través del reservorio Ommaya (vía eficaz para el tratamiento de la afectación meníngea). Es importante administrar el tratamiento quimioterápico con MTX29 antes de proceder al tratamiento radioterápico porque esta secuencia de administración minimiza los riesgos de leucoencefalopatía y neurotoxicidad tardía (deterioro cognitivo y ataxia) de la radioterapia asociada a MTX30. Se ha comprobado mayor deterioro cognitivo en los pacientes tratados con radioterapia y quimioterapia, que en aquéllos tratados únicamente con quimioterapia31. Dicha neurotoxicidad parece ser mayor en individuos mayores de 60 años (grupo de pacientes frecuentemente afectado por linfoma cerebral primario)32. Por ello, se están llevando a cabo ensayos clínicos de regímenes de poliquimioterapia complementarios al metotrexate a altas dosis33,34 (topotecan35, temozolamida, rituximab intratecal36...) sin radioterapia para estos pacientes37-39. Para los pacientes menores de 60 años, el tratamiento de consolidación de elección sería la radioterapia holocraneal39,40, ya que el retraso del tratamiento radioterápico podría conllevar un peor control de la enfermedad en estos pacientes más jóvenes32. La dosis estándar de radiación holocraneal es de 45 Gy. Aquellos pacientes con linfoma intraocular deben recibir entre 30 y 40 Gy de radiación intraocular37. Las dosis suelen ser menores que para otros tumores cerebrales. No se recomienda radiación profiláctica espinal, excepto en los raros casos de linfoma intramedular. Se ha descrito neurotoxicidad para dosis totales superiores a 36 Gy y dosis diarias fraccionadas superiores a 1,8 Gy40. Con todo las dosis suelen ser menores que para otros tumores cerebrales. Tres semanas tras concluir la radioterapia se instaura tratamiento quimioterápico con altas dosis de AraC iv.
El tratamiento del PCNSL plantea numerosas controversias en la actualidad, tales como el papel de la radioterapia y de la quimioterapia intratecal, la dosis y el momento de administración de la radioterapia y la protocolización de distintas pautas de tratamiento en función de la edad del paciente. Para resolver éstas y otras cuestiones se precisa ahondar en el estudio del PCNSL y la realización de potentes ensayos clínicos40,34
PRONÓSTICO
El pronóstico de los linfomas primarios de SNC es peor que el de los linfomas sistémicos41. La supervivencia media sin tratamiento es de 1,8-3,3 meses tras el diagnóstico. Con radioterapia, la supervivencia media es de 10 meses39 y con la adición de quimioterapia y MTX intratecal se consigue una tasa de respuesta inicial del 85% y las recurrencias suelen aparecer entre los 15 y 45 meses del tratamiento. En general, la supervivencia es mayor que en los gliomas de alto grado.
Entre los factores de mejor pronóstico se encuentran: lesión intracraneal única, ausencia de tumor meníngeo o periventricular, ausencia de inmunodeficiencia, edad menor de 60 años, Karnofsky superior a 70 y tratamiento radioterápico combinado con quimioterapia (antraciclinas y metotrexate). Se ha relacionado con peor pronóstico la resección quirúrgica parcial, los niveles séricos elevados de lactato deshidrogenasa, la hiperproteinorraquia y la afectación de estructuras cerebrales profundas42. No se ha demostrado valor pronóstico del subtipo histológico ni de los marcadores de proliferación. Se han relacionado las pérdidas en 6q con un peor pronóstico18. Se ha encontrado una correlación estadísticamente significativa entre la expresión del antígeno bcl-6 y una mayor supervivencia en pacientes afectos de PCNSL43. Ninguna de las clasificaciones existentes de los linfomas sistémicos incluye específicamente a los linfomas primarios del SNC y no existe una clasificación específica de los mismos. Una clasificación basada en características genéticas (marcadores pronósticos), podría tener utilidad clínica en cuanto a pronóstico y tratamiento44,45.
LINFOMA PRIMARIO DE SNC Y SIDA
Los linfomas primarios del sistema nervioso central (LPSNC) son tumores agresivos tanto en pacientes con SIDA como en personas no infectadas por el virus de la inmunodeficiencia humana tipo 1 (VIH-1). Entre 1980 y 1989 la frecuencia de estas neoplasias se incrementó 9 veces, con una incidencia absoluta de 4,7 casos/1.000 personas/año en pacientes con SIDA. Suelen aparecer en fases avanzadas de la enfermedad (CD4<200/µl, estadio C3). Estos enfermos tienen un riesgo 2.600 veces mayor de padecer esta complicación en comparación con la población general. A partir de la introducción del tratamiento antirretroviral de gran actividad (TARGA) en 1996, se ha modificado la historia natural de los linfomas no hodgkinianos (LNH) asociados con el SIDA, comprobándose una disminución en la incidencia de los LPSNC. Son tumores que muestran una fuerte asociación con el virus de Epstein-Barr (VEB) y representan el 15% de todos LNH que se observan en los pacientes con SIDA, en comparación con el 1% en aquellos no infectados por el VIH-1.
Su incidencia oscila entre el 3 y el 4% de los casos infectados por VIH y constituye la neoplasia que con mayor frecuencia compromete el SNC por lo que se la incluye entre las patologías marcadoras del SIDA. La edad media de aparición son 39 años y el 90% de los casos son varones. Se trata de tumores de gran malignidad y multicéntricos, que muestran predilección por la localización en ganglios basales, cuerpo calloso, tálamo, sustancia blanca periventricular y región subependimaria. Los tipos histológicos predominantes no difieren de los observados en los inmunocompetentes, predominando los tumores de células grandes, inmunoblásticos y centroblásticos, cuyas células contienen el genoma del VEB, lo cual no sucede en otros linfomas sistémicos asociados a SIDA.
Difieren en el diagnóstico, pronóstico y tratamiento respecto a los PCNSL en pacientes inmunocompetentes. Los linfomas primarios del SNC en pacientes con SIDA presentan más frecuentemente necrosis central y captación de contraste en anillo, lo que plantea el diagnóstico diferencial con toxoplasmosis cerebral46,47. La presencia de una lesión única sugiere más el diagnóstico de linfoma, mientras que la presencia de un número de lesiones superior a 4 hace que este diagnóstico sea más improbable. La toxoplasmosis cerebral se caracteriza por la presencia de una o más frecuentemente varias lesiones hipodensas que captan contraste en anillo y se acompañan de edema y de efecto masa. Las localizaciones más frecuentes son los ganglios de la base y los lóbulos frontoparietales. Aunque los hallazgos radiológicos de la toxoplasmosis cerebral son inespecíficos, la toxoplasmosis es la primera causa de lesiones ocupacionales intracraneales en pacientes con SIDA. La mayoría de los pacientes afectos de toxoplasmosis cerebral poseen serología positiva antitoxoplasma, siendo poco frecuente el diagnóstico de toxoplasmosis cerebral con serología negativa (menos del 20% de los casos). Aunque el diagnóstico definitivo de toxoplasmosis requiere la realización de una biopsia cerebral, ésta no está indicada como método diagnóstico inicial. En los centros que lo dispongan el PET-FDG48 (Positron Emision Tomography-fluorodeoxiglucosa), el Talio-201-SPECT (Single Photon Emision Computed Tomography) y la espectroscopia pueden ser útiles en el diagnóstico diferencial de lesiones cerebrales en pacientes con SIDA. Asimismo la detección mediante PCR (Polymerase Chain Reaction) diferencial de ADN de VEB en LCR de pacientes con SIDA tiene elevada sensibilidad y especificidad en el diagnóstico de linfoma primario del SNC en estos pacientes49,50. En todo paciente VIH (+) en el que se objetiven lesiones cerebrales múltiples que capten contraste el primer diagnóstico de presunción es de enfermedad por Toxoplasma gondii y el paciente debe recibir tratamiento empírico antitoxoplasma y dexametasona para el edema cerebral. La biopsia cerebral queda reservada para aquellos casos que no respondan al tratamiento empírico administrado durante 10-14 días, a no ser que otros estudios, como la PCR de LCR o el SPECT, mostrasen suficiente evidencia diagnóstica.
En los casos afectos de SIDA el pronóstico de los PCNSL es peor51,52. El tratamiento activo antirretroviral (TARGA) de la infección VIH ayuda a prolongar la supervivencia53. La supervivencia media sin tratamiento es de 3-6 meses y de 13 meses con tratamiento. Esta supervivencia acortadada se debe normalmente a infecciones oportunistas.
En conclusión, si bien la biopsia cerebral estereotáxica y el estudio histológico son necesarios para el diagnóstico definitivo de estos tumores y la implementación de una terapia adecuada, las técnicas no invasivas como la SPECT y la RM con espectroscopia pueden ayudar al diagnóstico precoz evitando el rápido deterioro clínico que caracteriza a estos pacientes. Del mismo modo, la fuerte asociación del VEB con la patogenia de estos tumores obliga a incluir la detección del genoma de este virus en LCR y tejido cerebral en el algoritmo de diagnóstico de las masas cerebrales ocupantes en pacientes con enfermedad VIH/SIDA avanzada.
CASUÍSTICA DE LINFOMAS CEREBRALES PRIMARIOS EN LA COMUNIDAD AUTÓNOMA DE NAVARRA EN LOS ÚLTIMOS 5 AÑOS (2000-2004)
Durante los últimos 5 años (2000-2004) se han diagnosticado 5 casos de linfoma cerebral primario en la Comunidad Autónoma de Navarra (aproximadamente 1 caso/500.000 habitantes/año). Todos los pacientes eran inmunocompetentes; 3 de ellos varones y 2 mujeres. La edad media en el momento del diagnóstico fue de 56,8 años. Tras el diagnóstico los pacientes fueron estadiados y tratados por el servicio de hematología. En ninguno de ellos se encontró linfoma sistémico en el momento del diagnóstico. En 3 casos la neuroimagen (RMN) mostró lesiones múltiples en el momento del diagnóstico y en 2 casos fueron lesiones únicas. En todos los casos las lesiones mostraron captación de contraste. En 2 de los casos además de lesiones supratentoriales se objetivaron lesiones a nivel infratentorial. En todos los casos hubo lesión/es supratentoriales, siendo la localización más frecuente periventricular a nivel temporal y frontal. En todos los casos las lesiones captaban contraste. Todos los pacientes debutaron con clínica neurológica (ninguno hallazgo casual), siendo la forma más frecuente (en 3 de los 5 casos) de debut la focalidad neurológica (hemiparesia, disfasia, parálisis de pares craneales VII y VIII, ataxia de la marcha, disartria de la marcha y dismetría). Otras formas de aparición clínica fueron; alteraciones cognitivas y de carácter en 2 de los 5 casos y crisis epilépticas en 1 de los 5 casos. Todos los pacientes fueron sometidos a biopsia estereotáctica excepto uno de ellos que fue sometido a resección tumoral por sospecha diagnóstica de astrocitoma y su supervivencia no ha resultado superior hasta el momento al resto de los casos únicamente biopsiados. Todos los casos fueron diagnósticados de linfoma no Hodgkin de células B, tratándose de un alto grado en 4 de los 5 casos y de bajo grado en el caso restante. Todos los pacientes recibieron tratamiento con metotrexate intratecal, radioterapia holocraneal y altas dosis de Ara-C basado en el protocolo DeAngelis. A todos ellos se les administró MTX intraventricular tras colocación de un reservorio Ommaya. Sólo uno de los casos requirió la retirada de este dispositivo debido a una infección del SNC. En la actualidad 2 pacientes han fallecido y 3 sobreviven. La supervivencia media tras el diagnóstico es en la actualidad de 25,6 meses. De los 3 pacientes que sobreviven en ninguno hay imágenes de recidiva en las últimas RMN de control practicadas. Todos ellos presentan cierto grado de deterioro cognitivo secundario a la radioterapia y en uno de los casos hay signos compatibles con radionecrosis en la última RMN practicada, que se correlaciona clínicamente con un deterioro cognitivo severo.
CONCLUSIONES
El PCNSL es un tipo de linfoma no Hodgkin poco frecuente, cuya incidencia ha sufrido un gran aumento en los últimos 30 años. Al contrario que los linfomas no Hodgkin sistémicos extranodales, tiene una pobre respuesta al tratamiento. Sin embargo, el cambio en los regímenes de tratamiento, basados en la quimioterapia con metotrexate y la radioterapia holocraneal, ha mejorado el pronóstico de estos pacientes. Desgraciadamente, la neurotoxicidad asociada a este tratamiento es muy elevada, especialmente en los pacientes mayores de 60 años. A pesar del progreso en el tratamiento de los PCNSL, existen aún numerosas controversias como la dosis y frecuencia óptimas de aplicación del metotrexate, el tratamiento de las recidivas54 y el establecimiento de protocolos de tratamiento más adecuados para cada edad. Estas cuestiones han propiciado la realización de diversos estudios clínicos buscando el modo de intensificar el tratamiento quimioterápico, limitar el uso de la radioterapia y disminuir así la potencial neurotoxicidad55. Los estudios de biología molecular buscan posibles oncogenes con valor pronóstico en el PCNSL, que ayudarían a desarrollar esquemas de tratamiento en función del riesgo de recidiva44.
Una clasificación basada en características genéticas (marcadores pronósticos), podría tener utilidad clínica en cuanto a pronóstico y tratamiento45.
BIBLIOGRAFÍA
1. Singh AD, Lewis H, Schachat AP. Primary lymphoma of the central nervous system. Ofthalmol Clin North Am 2005; 18: 199-207. [ Links ]
2. Batara JF, Grossman SA. Primary central nervous system lymphomas. Curr Opin Neurol 2003; 16: 671-675. [ Links ]
3. Hao D, DiFrancesco LM, Brasher P.M, deMetz C, Fulton DS, DeAngelis LM et al. Is primary CNS lymphoma really becoming more common? A population-based study of incidence, clinicopathological features and outcomes in Alberta from 1975 to 1996. Ann Oncol 1999; 10: 65-70. [ Links ]
4. Camilleri-Broet S, Davi F, Feuillard J, Seilhean D, Michiels JF, Brousset P et al. AIDS-related primary brain lymphomas: histopathologic and immunohistochemical study of 51 cases. The French study group for HIV-associated tumors. Hum Pathol 1997; 28: 367-374. [ Links ]
5. Levy-Clarke GA, Chan CC, Nussenblatt RB. Diagnosis and management of primary intraocular lymphoma. Hematol Oncol Clin North Am 2005; 19: 739-749. [ Links ]
6. Coulon A, Lafitte F, Hoang-Xuan K, Martin-Duverneuil N, Mokhtari K, Blustajn J et al. Radiographic findings in 37 cases of primary CNS lymphoma in immunocompetent patients. Eur Radiol 2002; 12: 329-340. [ Links ]
7. Erdag N, Bhorade RM, Alberico RA, Yousuf N, Patel MR. Primary lymphoma of the central nervous system: typical and atypical CT and MR imaging appearances. AJR Am J Roentgenol 2001; 176: 1319-1326. [ Links ]
8. Kuker W, Nagele T, Korfel A, Heckl S, Thiel E, Bamberg M et al. Primary central nervous system lymphomas (PCNSL): MRI features at presentation in 100 patients. J Neurooncol 2005; 72: 169-177. [ Links ]
9. Vaquero J, Martinez R, Rossi E, Lopez R. Primary Cerebral Lymphoma: the Ghost Tumor. J Neurosurg 1984; 60: 174-176. [ Links ]
10. Greenberg MS. Handbook of Neurosurgery (4th. Ed.). Greenberg Graphics, Lakeland, 1997; 294-297. [ Links ]
11. Howard A, Fine MD, Mayer RJ. Primary Central Nervous System Lymphoma. Ann Intern Med 1993; 119: 1093-1104. [ Links ]
12. Kleihues P, Cavenee WK. Tumours of the Nervous System. Pathology and Genetics. World Health Organization Classification of Tumours. IARC Press, Lyon, 2000; 198-203. [ Links ]
13. Shenkier TN, Blay JY, O´Neill BP, Poortmans P, Thiel E, Jahnke K et al. PCNSL of T-cell origin: a descriptive analysis from the international PCNSL collaborative group. J Clin Oncol 2005; 23: 2233-2239. [ Links ]
14. Jahnke K, Thiel E, Schiling A, Herrlinger U, Weller M, Coupland SE et al. Low-grade primary central nervous system lymphoma in immunocompetent patients. Br J Haematol 2005; 128: 616-624. [ Links ]
15. Guinto G, Felix I, Arechiga N, Arteaga V, Kovacs K. PCNSL in immunocompetent patients. Histol Histopathol 2004; 19: 963-972. [ Links ]
16. Nakamura M, Shimada K, Ishida E, Konishi N. Histopathology, patogénesis and molecular genetics in PCNSL. Histol Histopathol 2004; 19: 211-219. [ Links ]
17. Paulus W, Jellinger K, Morgello S. Malignant lymphomas. Pathology and Genetics of Tumours of the Nervous System. Ed. by Kleihuis and P. Cavenee WK. Lyon, France, International Agency for Research on Cancer 1997; 154-159. [ Links ]
18. Rickert CH, Dockhorn-Dworniczak B, Simon R, Paulus W. Chromosomal imbalances in Primary Lymphomas of the Central Nervous System. Am J Pathology 1999; 155: 1445-1451. [ Links ]
19. Ferreri AJ, Dell´Oro S, Capello D, Ponzoni M, Iuzzolino P, Rossi D et al. Aberrant methylation in the promoter region of the reduced folate carrier gene is a potencial mechanism of resistance to methotrexate in PCNSL. Br J Haematol 2004; 126:657-664. [ Links ]
20. Zhang SJ, Endo S, Saito T, Kuono M, Kuroiwa T, Washiyama K et al. Primary malignant lymphoma of the brain: frequent abnormalities and inactivation of p14 tumor suppressor gene. Cancer Sci 2005; 96: 38-41. [ Links ]
21. Julien S, Radosavljevic M, Labouret N, Camilleri-Broet S, Davi F, Raphael M et al. AIDS primary central nervous system lymphoma: molecular analysis of the expressed VH genes and possible implications for lymphomagenesis. J Immunol 1999; 162: 1551-1558. [ Links ]
22. Fitzsimmons A, Upchurch K, Batchelor T. Clinical features and diagnosis of PCNSL. Hematol Oncol Clin North Am 2005; 19: 689-703. [ Links ]
23. DeAngelis LM, Yahalom J, Thaler HT, Kher U. Combined Modality Therapy for Primary CNS Lymphomas. J Clin Oncol 1992; 10: 635-643. [ Links ]
24. Plotkin SR, Batchelor TT. Primary nervous system lymphoma. Lancet Oncol 2001; 2: 354-365. [ Links ]
25. Hoang-Xuan K, Camilleri-Broet S, Soussain C. Recent advances in primary CNS lymphoma. Curr Opin Oncol 2004; 16: 601-606. [ Links ]
26. Yamanaka R, Tanaka R. Advances for the treatment of primary central nervous system lymphoma (review). Oncol Rep 2004; 12: 563-568. [ Links ]
27. Shenkier TN, Blay JY, O´Neill BP, Poortmans P, Thiel E, Jahnke K et al. Primary CNS lymphoma of T-cell origin: a descriptive analysis from the international primary CNS lymphoma collaborative group. J Clin Oncol 2005; 23: 2233-2239. [ Links ]
28. DeAngelis LM, Yahalom J. Primary central nervous system lymphoma. En: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: principles and practice of oncology. Philadelphia: Lippincott-Raven, 1997: 2233-2242. [ Links ]
29. Batchelor TT, Carson K, ONeill A, Grossman SA, Alavi J, New P et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol 2003; 21: 1044-1049. [ Links ]
30. McAllister LD, Doolittle ND, Guastadisegni PE, Kraemer DF, Lacy CA, Crossen JR et al. Cognitive outcomes and long-term follow-up results after enhanced chemotherapy delivery for primary central nervous system lymphoma. Neurosurgery 2000; 46: 51-60. [ Links ]
31. Correa DD, DeAngelis LM, Shi W, Thaler H, Glass A, Abrey LE. Cognitive functions in survivors of primary central nervous system lymphoma. Neurology 2004; 62: 532-533. [ Links ]
32. Deangelis LM, Hormigo A. Treatment of primary central nervous system lymphoma. Semin Oncol 2004; 31: 684-692. [ Links ]
33. Jahnke K, Korfel A, Martus P, Weller M, Herrlinger U, Schmittel A et al: on the behalf of the German Primary central Nervous System Lymphoma Study Group (G-PCNSL-SG). High-dose methotrexae toxicity in elderly patients with primary central nervous system lymphoma. Ann Oncol 2005; 16: 445-449. [ Links ]
34. Hoang-Xuan K, Camilleri-Broet S, Soussain C. Recent advances in primary CNS lymphoma. Curr Opin Oncol 2004; 16: 601-606. [ Links ]
35. Fisher L, Thiel E, Klasen HA, Kirchen H, Jahnke K, Korfel A. Response of relapsed or refractory PCNSL to topotecan. Neurology 2004; 62: 1885-1887. [ Links ]
36. Enting RH, Demopoulos A, DeAngelis LM, Abrey LE. Salvage therapy for PCNSL with a combination of rituximab and temozolamide. Neurology 2005; 64: 934. [ Links ]
37. Lauren A, Yahalom J, DeAngelis LM. Treatment for primary CNS lymphoma: the next step. J Clin Oncol 2000; 18: 3144-3150. [ Links ]
38. Singh AD, Lewis H, Schachat AP. Primary lymphoma of the central nervous system. Ofthalmol Clin North Am 2005; 18: 199-207. [ Links ]
39. Nguyen PL, Chakravarti A, Finkelstein DM, Hochberg FH, Batchelor TT, Loeffler JS. Results of whole-brain radiation as salvage of methotrexate failure for immunocompetent patients with primary CNS lymphoma. J Clin Oncol 2005; 23: 1507-1513. [ Links ]
40. Herrlinger U, Korfel A, Hebart H, Schlegel U, Plasswilm L, Illerhaus G et al. Meeting report: primary central nervous system lymphoma: standards of care and future perspectives. Onkologie 2005; 28: 109-111. [ Links ]
41. Kasalon YL, Ambinder RF. AIDS-related PCNSL. Hematol-Oncol-Clin North Am 2005; 19: 665-687. [ Links ]
42. Ferreri AJM, Blay J-Y, Reni M, Pasini F, Spina M, Ambrosetti A et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol 2003; 21: 266-272. [ Links ]
43. Braaten KM, Betensky RA, de Leval L, Okada Y, Hochberg FH, Louis DN et al. BCL-6 expression predicts improved survival in patients with primary central nervous system lymphoma. Clin Cancer Res 2003; 9: 1063-1069. [ Links ]
44. Ferreri AJ, Reni M. Establishing a prognostic score for primary CNS lymphomas. Int J Radiat Oncol Biol Phys 2005; 61: 303-304. [ Links ]
45. Yamanaka R, Akutagawa S, Taguchi F, Yajima N, Tsuchiya N, Uzuka T et al. selection of surrogate marker genes in PCNSL for radio-chemotherapy by DNA array analysis. Int J Oncol 2003; 23: 913-923. [ Links ]
46. Ruiz A, Post MJD, Bundschu C, Ganz WI, Georgiou M. Primary central nervous system lymphoma in patients with AIDS. Neuroimaging Clin N Am 1997; 7: 281-296. [ Links ]
47. Berger JR. Mass lesions of the brain in AIDS: the dilemmas of distinguishing toxoplasmosis from primary CNS lymphoma. AJNR Am J Neuroradiol 2003; 24: 554-555. [ Links ]
48. Hoffman JM, Waskin HA, Schiffer T, Hanson MW, Gray L, Rosenfeld S et al. FDG-PET in differenciating lymphoma from nonmalignant central nervous system lesions in patients with AIDS. J Nucl Med 1993; 34: 567-575. [ Links ]
49. Antinori A, De Rossi G, Ammassari A, Cingolani A, Murri R, Di Giuda D et al. Value of combined approach with thallium-201 single-photon emission computed tomography and Epstein-Barr virus DNA polymerase chain reaction in CSF for the diagnosis of AIDS-related primary CNS lymphoma. J Clin Oncol 1999; 17: 554-560. [ Links ]
50. Ivers LC, Kim AY, Sax PE. Predictive value of PCR of CSF for detection of EBV to establish the diagnosis of HIV-related PCNSL. Clin Infect Dis 2004; 38: 1629-1632. [ Links ]
51. Corti M, Villafone F, Trione N, Schtirbu R, Yampolsky C, Narbaitz M. PCNSL in AIDS patients. Enferm Infecc Microbiol Clin 2004; 22: 313-314. [ Links ]
52. Newell ME, Hoy JF, Cooper SG, DeGraaff B, Grulich AE, Bryant M et al. HIV-related PCNSL: factors influencing survival in 111 patients. Cancer 2004; 100: 2627-2636. [ Links ]
53. Skiest DJ, Crosby C. Survival is prolonged by highly active antiretroviral therapy in AIDS patients with PCNSL. AIDS 2003; 17: 1787-1793. [ Links ]
54. Plotkin SR, Betensky RA, Hochberg FH, Grossman SA, Lesser GJ, Nabors LB et al. Treatment of relapsed PCNSL with high-dose methotrexate. Clin Cancer Res 2004; 10: 5643-5646. [ Links ]
55. Gavrilovic IT, Abrey LE. Primary central nervous system lymphoma. Curr Oncol Rep 2004; 6: 388-395. [ Links ]

