










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.31 supl.3 Pamplona 2008
Enfermedades articulares y uveítis
Articular diseases and uveitis
J.M. Benítez del Castillo, D. Díaz-Valle, E. Pato, C. López Abad, N. Alejandre
Unidad de Superficie e Inflamación ocular. Hospital Clínico San Carlos. Madrid.
Dirección para correspondencia
RESUMEN
La inflamación ocular es una manifestación clínica frecuente de múltiples enfermedades sistémicas autoinmunes, siendo de gran relevancia en las espondiloartropatías. Dentro del grupo de las espondiloartropatías existen diferentes entidades clínicas, asociándose a diferentes patrones de uveítis. Se han definido una serie de patrones discriminativos que relacionan formas concretas de uveítis con determinadas enfermedades sistémicas u oculares. La uveítis anterior aguda unilateral recidivante es la más frecuente en las espondiloartropatías, y puede ser la forma de inicio de una espondiloartropatía no diagnosticada previamente. La colaboración entre oftalmólogos y reumatólogos o internistas es fundamental para el correcto manejo y tratamiento de estos pacientes.
Palabras clave: Uveítis. Espondiloatropatías. Artritis reumatoide. Enfermedad inflamatoria intestinal.
ABSTRACT
Ocular inflammation is a common clinical manifestation related to several autoimmune systemic disorders, specially spondyloarthropaties. In this group, there are different clinical diseases that are related to special uveitic patterns. Several discriminative patterns have been defined that closely link uveitis with certain systemic or ophthalmic diseases. Unilateral recurrent anterior acute uveitis is the most frequent form of uveitis related to spondyloarthropaties, and is sometimes the initial manifestation of an undiagnosed spondyloarthropaty. The collaboration of ophthalmologists, rheumatologists and internal medicine specialists is very important for the correct management and treatment of these patients.
Key words: Uveitis. Spondyloarthropaties. Rheumatoid arthritis. Inflammatory bowel disease.
Introducción
Las uveítis anteriores son las uveítis más frecuentes (3/4 partes del total de las mismas). La incidencia es de 8/100.000 habitantes. Aunque sean más fáciles de manejar, pueden llevar a la ceguera como consecuencia de dos complicaciones: glaucoma y edema macular1. Las espondiloartopatías constituyen una de las causas más frecuentes de uveítis anteriores. Las uveítis asociadas a espondiloartropatías se encuadran dentro del grupo de las uveítis relacionadas con el antígeno de histocompatibilidad HLA-B27. Dicho antígeno predispone a padecer uveítis anterior independientemente de posibilitar una espondiloartropatía. Dentro de las enfermedades relacionadas con el HLA-B27 y uveítis se definen la espondilitis anquilosante, el síndrome de Reiter, artritis psoriásica y la enfermedad inflamatoria intestinal (EII)2.
Las uveítis relacionada con el haplotipo HLA-B27 constituyen una entidad única3. Se trata de una uveítis anterior, aguda, recidivante y unilateral en cada brote4. En comparación con las uveítis anteriores crónicas son menos graves debido a la no constancia del proceso inflamatorio; no obstante, debido a su curso en brotes con gran actividad inflamatoria son severas en relación a otras uveítis agudas. Los afectados son con frecuencia varones y de edad ligeramente inferior que los pacientes con uveítis HLA- B27 negativas. Por todo ello, ante uveítis anteriores recidivantes solicitamos el tipaje HLA-B27 debido a su factor pronóstico. El hallazgo del haplotipo HLA-B27 tiene interés ya que indica la posible patogenia de la enfermedad. Las bacterias Gram negativas interactúan con los antígenos HLA clase I en el desencadenamiento de la uveítis anterior. Sin embargo, el tratamiento antibiótico no mejora ni disminuye el número de recidivas. Nosotros hemos observado que los pacientes con uveítis anterior con y sin sacroileítis asociada presentan infiltrados inflamatorios intestinales no sintomáticos5. En estos casos el tratamiento con antiinflamatorios intestinales tipo sulfasalazina reduce el número de brotes y la intensidad de éstos6.
En este trabajo se revisarán las uveítis asociadas a las espondiloartropatías seronegativas (espondilitis anquilosante, artropatía psoriásica, artritis reactiva o síndrome de Reiter y EII), así como la afectación ocular en el contexto de la artritis reumatoide (AR) del adulto y la artritis idiopática juvenil (AIJ).
Espondilitis anquilosante
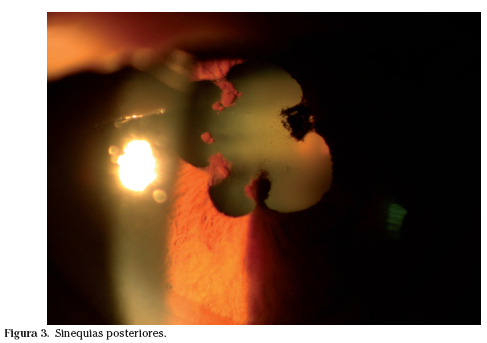
Los pacientes con espondilitis anquilosante (EA) padecen a lo largo de su enfermedad uveítis en el 25% de los casos. También se han descrito conjuntivitis asociada a la EA. En un número no desdeñable de casos la uveítis es el primer síntoma de la enfermedad y en muchos más ayuda a filiar una lumbalgia escasamente sintomática. Los dos ojos se afectan en el 80% de los casos aunque muy raramente se inflaman ambos a la vez. Por ello decimos que es una uveítis unilateral, entendiendo como tal en cada brote, aunque los brotes pueden afectar a ambos ojos en momentos diferentes. Se trata de una uveítis anterior, es decir una iritis o iridoclitis. Su curso es agudo, con dolor, ojo rojo, fotofobia y disminución de visión. Como consecuencia de la inflamación se rompe la permeabilidad de la barrera hemato-acuosa y la cámara anterior se llena de proteínas y células. Mediante el uso de la lámpara de hendidura (biomicroscopía) se puede observar la turbidez del humor acuoso por el exceso de proteínas y las células. En ocasiones, el número de células es tan grande que se depositan, dando lugar a un hipopion (Fig. 1). Las células también se pegan a la cara posterior de la córnea originando acúmulos denominados precipitados retroqueráticos (Fig. 2). Es recurrente; algunos pacientes pueden tener hasta un brote mensual. Característicamente los pacientes que ya han tenido varios brotes perciben la proximidad de una recidiva hasta 24 horas antes, cuando el examen con la lámpara de hendidura es aún absolutamente normal7. El empleo del láser flaremeter permite detectar mínimas alteraciones de la permeabilidad de la barrera hemato-acuosa antes y después de la normalización biomicroscópica de la misma. La disminución de visión que tiene lugar durante el ataque se debe fundamentalmente a la turbidez del humor acuoso y ocasionalmente a un edema macular secundario. Como consecuencia del proceso inflamatorio el iris tiende a pegarse al cristalino (sinequias posteriores) (Fig. 3) y a la periferia corneal (sinequias anteriores).


La EA ocurre más frecuentemente en varones, unas 2,5-3 veces más que en mujeres8. Las mujeres suelen tener una forma más leve y con afectación articular más periférica. Afecta al 0,1% de los adultos de raza blanca9. El 96% de los pacientes con EA tiene el HLA-B27 positivo, mientras que sólo lo es en el 6-14% de los adultos blancos sanos. No obstante, sólo el 1,3% de los pacientes HLA-B27 positivos desarrollan EA. La manifestación principal de la enfermedad es la sacroileítis. Ésta suele ser asintomática al principio de la enfermedad. Con el tiempo aparece dolor en la parte baja de la espalda y rigidez sobre todo matutina ya que aumenta con la inactividad. Las radiografías de las articulaciones sacroilíacas demuestran un borramiento y esclerosis de las mismas. La tomografia computarizada puede detectar cambios patológicos en casos radiográficamente normales. Si la enfermedad progresa se fusiona toda la columna, imposibilitando el movimiento y originando ocasionalmente problemas respiratorios. Otras alteraciones relacionadas con la enfermedad son: insuficiencia aórtica y problemas en la conducción cardiaca. La uveítis no se correlaciona con la gravedad de la EA.
Es importante diagnosticar la enfermedad en sus estadios iniciales para evitar la deformidad de la columna. El tratamiento se basa en ejercicios y uso de antiinflamatorios no esteroideos. En casos graves el empleo de fármacos con acción antiTNF resulta útil. La afectación ocular requiere la administración de corticoides y midriáticos tópicos en una pauta intensiva al principio hasta controlar la respuesta inflamatoria y un descenso gradual durante al menos 4-6 semanas. Algunos pacientes puden precisar una inyección periocular de corticoides y es excepcional que sea necesario administrar corticoides por vía sistémica en estos casos.
Síndrome de reiter
El síndrome de Reiter (SR) es una enfermedad caracterizada por la triada artritis, uretritis y conjuntivitis. Afecta a varones jóvenes y también se asocia al HLA-B27. La enfermedad aparece en el 1% de los pacientes que padecen una uretritis no gonocócica (Clamidia o Ureaplasma urelítico) y en el 2% de los que sufren una infección intestinal por Shigella10. Otras manifestaciones sistémicas que pueden ocurrir en el SR son: balanitis, queratoderma blenorrágico (semejante a psoriasis) y aftas orales (no confundir con las de la enfermedad de Behçet)11. Las manifestaciones articulares incluyen: tenosinovitis, fascitis plantar, sacroileítis y oligoartropatías inflamatorias (manos, muñecas, pies y rodillas). La mayoría de los pacientes desarrollan signos 2-4 semanas después de la infección. No obstante, el cuadro completo puede tardar años en aparecer.
La conjuntivitis aparece en el 30-60% de los pacientes. Más rara es la queratitis, en forma de infiltrados subepiteliales. La iridociclitis aparece en el 3-12% de los casos. La uveítis es semejante a la observada en los pacientes con EA.
El tratamiento de la infección inicial no parece alterar el curso del SR. El tratamiento del SR incluye el empleo de antiinflamatorios y ocasionalmente inmunosupresores.
Artropatía psoriásica
Hasta un 30% de los pacientes con artritis psoriásica tienen afectación ocular. En el 20% de los casos se presenta en forma de conjuntivitis, y sólo en un 7-10% se manifiesta como uveítis anterior aguda recidivante unilateral. Un pequeño porcentaje de los casos de uveítis puede tener una evolución crónica, o bien presentarse en forma de uveítis anterior aguda bilateral. Es más frecuente la afectación ocular en los pacientes de sexo masculino, en los que la psoriasis afecta el cuero cabelludo o las zonas ciliares, y en los que presentan artritis, sobre todo sacroileítis y espondilitis.
Dado que la psoriasis es una enfermedad muy común, no debe atribuirse automáticamente a ésta cualquier uveítis que aparece en estos pacientes, especialmente si no existe artritis asociada o el patrón de afectación ocular no es característico. El pronóstico ocular es, en general, muy bueno. El tratamiento de las manifestaciones cutáneas de la enfermedad no altera el curso de la enfermedad ocular.
Enfermedad inflamatoria intestinal
La enfermedad inflamatoria intestinal (EII) es un trastorno crónico de etiología desconocida que abarca dos entidades: la Colitis Ulcerosa (CU) y la Enfermedad de Crohn (EC). La incidencia de éstas es del 10.4 y el 5.6 por 100.000 habitantes/año respectivamente, aunque la de la EC parece en aumento12.
Aunque su etiología es desconocida parece que se debe a la acción de un desencadenante externo sobre un sistema inmune predispuesto. Este desencadenante entraría en contacto con el sistema inmune a través del tubo digestivo. En este sentido se han encontrado asociaciones con algunos tipos de HLA y un aumento de la prevalencia entre los familiares de primer grado13, así como algunos trabajos que relacionan la aparición de la enfermedad con el Mycobacterium paratuberculosis14. El sistema inmune activado produce las lesiones digestivas liberando además al resto del organismo citoquinas proinflamatorias que producen una acción directa en diversos órganos además de activar la cascada del complemento. De esta manera la enfermedad produce un daño de tipo vasculítico-pretrombótico que puede afectar a numerosas regiones del organismo15.
Manifestaciones clínicas
La EC puede afectar cualquier parte del tracto gastrointestinal, siendo el íleo terminal la zona más fecuentemente afectada. En la colitis ulcerosa, alrededor del 55% de los pacientes tienen proctitis, un 30% colitis izquierda y un 15% pancolitis. Es característico que la EC tenga un aspecto de empedrado al presentar zonas de mucosa aparentemente sana alternando con zonas afectadas, mientras que la CU no tiene ese aspecto segmentado.
Entre las manifestaciones extraintestinales más frecuentes se encuentran la artritis, la fiebre, la pérdida de peso (muchas veces con déficit de B12 y litiasis biliar, cuando la EC afecta al ileon terminal), la aparición de eritemas (rash) y el Pyoderma gangrenosum16.
Manifestaciones oculares
La incidencia de manifestaciones oculares oscila entre el 3,5 y el 11,8%. Hasta un 68% de ellos presentará además otra manifestación extraintestinal, sobre todo artritis y espondilitis anquilosante. Aunque las complicaciones oculares suelen aparecer en pacientes ya diagnosticados de EII, pueden ser también la manifestación inicial. Su aparición suele implicar que la EII está activa aunque también podría no estarlo. Se ha publicado que la enfermedad ocular no depende de la extensión de la afectación intestinal y que suele aparecer en los primeros años.
La epiescleritis aparece hasta en un 29% de los pacientes con EII. Puede ser difusa o nodular, uni o bilateral, y se relaciona con la actividad intestinal, resolviéndose con el tratamiento de ésta.
La escleritis aparece en un 18% y también puede ser nodular o difusa. Las recurrencias pueden llevar a la escleromalacia, pero el pronóstico es bueno con un buen control de la EII. Si afecta la zona perilímbica puede producir estrías limbares y/o infiltrados corneales.
La afectación corneal es rara y suele ser secundaria a una escleritis en forma de infiltrados nodulares subepiteliales. Éstos pueden coalescer y producir una vascularización secundaria. Además, la inflamación puede adelgazar esa zona de la córnea con el consiguiente astigmatismo.
El ojo seco y la queratitis aunque acompañan a muchas enfermedades autoinmunes, no suelen aparecer en la EII.
Hasta en el 17% de los pacientes puede aparecer uveítis, siendo algo más frecuente en mujeres. En muchas ocasiones se trata de episodios autolimitados de uveítis anteriores crónicas de inicio insidioso, pero no son uveítis silentes, por lo que las revisiones periódicas en general no son necesarias. La afectación posterior en forma de coriorretinitis es relativamente frecuente, existe una serie en la que aparece en un 10% de los pacientes con EII. Formas más raras parecidas a la epiteliopatía pigmentaria placoide multifocal posterior aguda también han sido descritas.
Aunque raras, han sido descritas las vasculitis retinianas con oclusión de ramas arteriales o venosas e incluso de la arteria o la vena central de la retina. También pueden aparecer miositis o pseudotumores orbitarios, con buena respuesta a esteroides sistémicos, pero de fácil recurrencia.
El 4% de las complicaciones oculares de la EII son neuritis ópticas anteriores vasculíticas, aunque neuritis retrobulbares y neurorretinitis también han sido descritas, aisladas o acompañando a otros cuadros oculares17.
El tratamiento de la inflamación ocular se basa en la administración de midriáticos y corticoides tópicos, perioculares o sistémicos en función del patrón de afectación, así como en el control de la enfermedad de base.
Artritis reumatoide
Entre un 20 y un 35% de los pacientes con artritis reumatoide (AR) pueden desarrollar diferentes manifestaciones oculares en el curso de su enfermedad. En la mayoría de los casos aparecen en las fases avanzadas de la misma. La queratoconjuntivitis seca es el tipo de afectación ocular más frecuente en la AR, seguida de la episcleritis, la escleritis y la afectación corneal. La AR no se incluye en el diagnóstico diferencial de ningún tipo de uveítis y debe considerarse que en un paciente con AR y uveítis la causa de la uveítis debe ser otra18.
Queratoconjuntivitis seca
La queratoconjuntivitis seca (QCS) es la manifestación ocular más frecuente en la AR. Entre un 10 y un 35% de los pacientes con AR tienen un ojo seco.
En la patogenia de la QCS la glándula lacrimal es infiltrada por linfocitos T y B, produciendo una atrofia secundaria de la glándula que es la responsable de la disminución en la producción de lágrima, o del deterioro de la calidad de la misma.
Los síntomas típicos son irritación, escozor, sensación de cuerpo extraño y fotofobia que empeoran sobre todo en ambientes secos y al final del día. Los enfermos tienen una mayor predisposición a padecer conjuntivitis infecciosas, úlceras corneales y perforación corneal.
El diagnóstico se realiza por el test de Schrimer y el empleo de tinciones córneo-conjuntivales. El principal problema es que resulta poco fiable en ocasiones y es relativamente molesto para el paciente.
En general, el tratamiento de la sequedad ocular y de la QCS es poco satisfactorio en los casos en los que la disminución de la producción de lágrimas es severa. El tratamiento básico es el uso de lágrimas artificiales. Actualmente, está aprobado por la FDA el uso de ciclosporina A en solución oftálmica al 0,05% en la QCS del síndrome de Sjögren, con beneficio objetivo y subjetivo en los pacientes19.
Episcleritis
La episcleritis representa una afectación ocular más superficial y leve que la escleritis. Es habitualmente autolimitada, aunque con frecuencia recidiva. Suele tratarse de formas difusas y habitualmente no precisan tratamiento sistémico. Se controla fácilmente con corticoides tópicos y en los casos con riesgo de complicaciones oculares y/o mala respuesta al tratamiento tópico se pueden utilizar antiinflamatorios no esteroideos o corticoides por vía oral.
Escleritis
La escleritis es una vasculitis de los vasos de la esclera. Menos de un 5% de los pacientes diagnosticados de AR desarrollarán una escleritis en su evolución. Sin embargo, esta cifra aumenta en los pacientes de más larga evolución y mayor agresividad. Clínicamente se caracterizan por edema e inflamación en la esclera. Sus síntomas principales son el dolor y el enrojecimiento localizado en uno de los cuadrantes del ojo. La escleritis asociada a AR suele ser anterior, y en un 60% de los casos es bilateral. La AR puede asociarse con cualquier forma de escleritis, aunque son más frecuentes las formas difusas y necrosantes que las nodulares. Las formas difusas son las más leves; y son más graves las nodulares y, sobre todo, las necrotizantes que pueden progresar hacia la perforación ocular.
Aunque puede considerarse rara, la complicación más temida en estos enfermos es la escleromalacia perforante. En estos casos es necesario un tratamiento quirúrgico.
Los antiinflamatorios no esteroideos constituyen el tratamiento inicial de elección en pacientes con AR y escleritis difusa o nodular. Cuando el tratamiento inicial fracasa o en pacientes con escleritis necrosante se debe recurrir a la administración de corticoides (a dosis entre 20 y 60 mg/día de prednisona o equivalente), con disminución progresiva de la dosis hasta suspenderlos o alcanzar una dosis baja de mantenimiento. En los casos en los que son necesarias dosis inaceptablemente altas de corticoides o es necesaria su administración durante mucho tiempo se pueden utilizar inmunosupresores como el metotrexato, la ciclofosfamida, la azatioprina, o la ciclosporina A , a las dosis habituales. La existencia de una escleritis necrosante es la indicación de utilizar inmunosupresores precozmente desde el inicio para muchos autores.
Afectación corneal
En un paciente con AR puede existir afectación corneal como: a) complicación de una queratoconjuntivitis seca; b) complicación de una escleritis o, más raramente, una episcleritis y c) sola, sin otros trastornos oculares. Las formas más frecuentes son la ulceración o el adelgazamiento corneal periférico (Fig. 4), la queratolisis (o melting corneal) y la queratitis esclerosante.

Clínicamente suelen aparecer de forma brusca, con dolor intenso y una cierta disminución de la agudeza visual. El diagnóstico diferencial se establece con cuadros de queratitis infecciosa y, dado que el tratamiento suele ser agresivo, es obligatorio descartar siempre la existencia de infección primaria o concomitante en el ojo. El pronóstico ocular es muy malo debido a la alta morbilidad ocular y su mala respuesta al tratamiento convencional.
La queratolisis (melting) es una complicación rara caracterizada por un «derretimiento» o adelgazamiento de la córnea que puede evolucionar a la ulceración o perforación ocular. La queratitis esclerosante es una complicación de las escleritis que se produce como resultado de la extensión del proceso inflamatorio con formación de neovasos y opacificación de la córnea.
Los principales problemas de la afectación corneal en la AR son su aparición en pacientes con la enfermedad bien controlada y la pobre respuesta al tratamiento. De hecho, los problemas corneales son la causa más frecuente de perforación ocular en pacientes con AR. Con mucha frecuencia el tratamiento con dosis altas de corticoides puede ser insuficiente, evolucionando el ojo hacia la perforación. Por este motivo se ha postulado iniciar el tratamiento concomitantemente con corticoides e inmunosupresores, lo cual parece mejorar el pronóstico ocular. La ciclosporina A parece efectiva en el tratamiento de algunos de estos pacientes; también el tratamiento con ciclofosfamida parece ser muy efectivo. En los casos en los que la perforación ocular parece inminente, se debe plantear la cirugía.
Artritis idiopática juvenil
La artritis indiopática juvenil (AIJ) es la enfermedad inflamatoria del tejido conectivo más frecuente en la infancia. Engloba todas aquellas artritis de causa desconocida que aparecen en menores de 16 años con una duración de más de 6 semanas. Es la causante del mayor grado de invalidez en niños20.
Su origen es desconocido aunque existe cierta predisposición genética sobre la que inciden una serie de factores desencadenantes. Todas ellas tienen en común la presencia de artritis (se considera el hallazgo fundamental para el diagnóstico), rash cutáneo y fiebre de carácter intermitente acompañada de otros signos (lifoadenopatías, hepatoesplenomegalia) y alteraciones analíticas (leucocitosis, anemia, VSG elevada) que hacen confundir el diagnóstico, sobre todo si la artritis no es evidente al inicio de la enfermedad.
Actualmente se recomienda la clasificación realizada por el American Collegue of Reumatology21-22 que se expone en la tabla 1.

La AIJ se puede confundir con una serie de entidades por lo que debemos hacer diagnóstico diferencial con:
1. Artritis post-infecciosa23.
2. Artritis reactiva asociada a infecciones bacterianas.
3. Otras enfermedades del tejido conectivo (fiebre y artritis son también manifestaciones del lupus, enfermedad mixta del tejido conectivo, dermatomiositis, sarcoidosis, etc.).
4. Procesos neoplásicos: leucemias, linfomas u otros procesos neoplásicos.
La comorbilidad de la AIJ está determinada, por lo general, por la presencia de uveítis de carácter crónico, no granulomatosa y/o recidivante en aproximadamente un 20% de los casos24. La mayor parte de los brotes ocurren durante el primer año de evolución de la AIJ, pero incluso 5 años después de la última evidencia de actividad articular de la enfermedad se pueden presentar episodios de uveítis.
Uveítis
Es la complicación más seria de la AIJ de inicio oligoarticular, con una incidencia de hasta un 20%25. El subgrupo de pacientes de sexo femenino con ANA positivos presenta un mayor riesgo de desarrollar algún episodio de uveítis. De todos modos, la negatividad de los ANA no excluye la posibilidad de poder desarrollar esta complicación.
En la AIJ, la forma de uveítis más frecuente es la uveítis anterior de curso crónico que puede ser unilateral al inicio, pero que en dos tercios de estos niños se hace bilateral a lo largo del primer año. Los signos clínicos, que no siempre están presentes, más frecuentes son: hiperemia ciliar, precipitados retroqueráticos, células inflamatorias en cámara anterior (Tyndall positivo), sinequias anteriores (puntos de unión entre iris y ángulo camerular) y sinequias posteriores (uniones entre iris y cristalino) con irregularidad del borde pupilar. Pueden presentar aumento de presión intraocular e inflamación del vítreo anterior. La exploración oftalmológica básica incluye: agudeza visual, biomicroscopía, presión intraocular y oftalmoscopía. A veces, son necesarias exploraciones adicionales como tomografía de coherencia óptica (OCT), ecografía ocular, angiografía y/o retinografía.
Son de difícil diagnóstico porque el niño es habitualmente asintomático (no se queja de dolor ni de pérdida de agudeza visual), los signos clínicos de uveítis son inaparentes en la mayoría de los casos al comienzo de la misma, y por la mala colaboración existente a la hora de la exploración. También son de difícil tratamiento por la dificultad de aplicar determinados tratamientos oculares (colirios, inyecciones peribulbares, láser...), y sistémicos (efectos secundarios ligados a muchos de ellos).
Tiene peor pronóstico que en los adultos ya que el 25-35% de los casos llegan a tener pérdidas de agudeza visual severas con alto grado de ambliopía al presentarse en niños menores de 7 años y con el desarrollo visual aún incompleto. El 30-40% de los niños tienen complicaciones oftalmológicas secundarias a la uveítis en el momento del diagnóstico (35-66% catarata, 25-55% queratopatía en banda, 10-20% ambliopía, 10-42% glaucoma, 7-42% edema macular, 4-17% ptisis bulbi). Todo ello obliga a realizar revisiones multidisciplinarias entre reumatólogo infantil, oftalmólogo y pediatra y a seguir unas guías con protocolos de actuación26 que la Academia Americana de Reumatología recomienda en función de la duración de la enfermedad, positividad de ANA, y el riesgo estimado del niño de padecer uveítis27.
El protocolo de seguimiento oftalmológico, tengan o no uveítis es:
Año de inicio menor de 6 años y ANA +
– Duración de la enfermedad < 4 años: evaluación ocular cada 3 meses.
– Duración de la enfermedad > 4 años: evaluación ocular cada 6 meses.
– Duración de la enfermedad > 7 años: evaluación ocular cada 12 meses.
Año de inicio mayor de 6 años y ANA +
– Duración de la enfermedad < 4 años: evaluación ocular cada 6 meses.
– Duración de la enfermedad > 4 años: evaluación ocular cada 12 meses.
Año de inicio menor de 6 años y ANA -
– Duración de la enfermedad < 4 años: evaluación ocular cada 6 meses.
– Duración de la enfermedad > 4 años: evaluación ocular cada 12 meses.
Año de inicio mayor de 6 años y ANA -
– Evaluación ocular cada 6 meses.
Siguiendo el esquema anterior, una vez el oftalmólogo haya realizado el diagnóstico de uveítis, la respuesta al tratamiento pautado así como la presencia de complicaciones determinará la frecuencia de las revisiones oftalmológicas.
Existen unos factores de riesgo de pérdida visual que son: comienzo temprano de la uveítis (especialmente si es antes que la artritis), ANA positivos, afectación bilateral, género femenino y presencia de complicaciones con baja agudeza visual al diagnóstico.
Los resultados visuales en uveítis ligada a AIJ aun siguiendo un protocolo de tratamiento y seguimiento estricto son: el 40% de los niños empeora su AV, el 20% acaban con baja AV y el 12% acaban con ceguera funcional (en los casos de enfermedad oligoarticular por inflamación crónica persistente). Según estos porcentajes no debemos tolerar inflamación persistente, incluso de bajo grado, durante periodos prolongados. Esto nos obliga a seguir con rigor las guías de revisiones recomendadas aunque no se observen signos o síntomas de afectación ocular28.
Tratamiento de la uveítis asociada a AIJ
Sin factores de riesgo de pérdida visual
El tratamiento lo constituyen corticoides tópicos en régimen intenso desde el principio29 y descenso lento y progresivo durante varias semanas asociados a midriáticos/ciclopléjicos mientras exista inflamación. El 20% de las uveítis en niños no responden al tratamiento tópico o responden al principio, pero al ir disminuyendo la dosis vuelven a activarse, por lo que será necesario añadir otros tratamientos como: inyecciones subtenonianas en casos unilaterales con gran inflamación anterior o, vitritis intensa, edema macular o hipotonía y/o AINE sistémicos en cuadros de inflamación crónica / severa. Los AINE tópicos no son efectivos por sí solos por lo que se utilizan de mantenimiento.
Con factores de riesgo de pérdida visual o alto grado de inflamación
– Primer escalón: corticoides sistémicos. A dosis de 1-2 mgs/Kg de peso/día o pulsos de metilprednisolona de 10-20 mgs/Kg/día durante 3 días. No se recomiendan tratamientos prolongados por los efectos secundarios sobre todo en el desarrollo30.
– Segundo escalón: inmunosupresores. Existe un grupo de recomendaciones realizadas por un panel de expertos para el uso de estos fármacos en las uveítis refractarias31. Se utilizan en niños con factores de riesgo de pérdida visual o aun sin factores de riesgo de pérdida visual cuando el proceso no está controlado en 4-6 semanas o bien antes si se requiere, si se ha reactivado con dosis bajas de corticoides o si queremos asociarlos a corticoides para reducir efectos secundarios.
Los inmunosupresores modificadores de la enfermedad más utilizados son:
– Metotrexate. Es la droga de elección en los niños por su seguridad, buena tolerancia y fácil administración. Ha demostrado su eficacia tanto para la afectación articular como para la uveítis32-33. Se pauta 10-15 mg/m2 en dosis única semanal, por vía oral o subcutánea. Usado en monoterapia puede administrarse hasta 20-25 mg/m2 por vía subcutánea (en la población infantil las dosis altas son bien toleradas incluso con mayor eficacia34).
– Ciclosporina A. Produce buenos resultados en alguna serie de AIJ y uveítis, pero con efectos secundarios. La dosis inicial es 3-5 mg /kg/día en dos tomas. Se usa combinada con metotrexate o como alternativa al mismo, si hay intolerancia o mala respuesta. Exige controles renales y de presión arterial cada 3 meses35.
– Micofenolato mofetilo. Disminuye las recidivas durante el tratamiento, así como la dosis de corticoides y presenta escasos efectos secundarios. La pauta utilizada es de 500 mg/12 horas por vía oral la primera semana hasta 2 gramos diarios de mantenimiento36.
– Azatioprina y ciclofosfamida. Se desaconseja su uso actualmente37.
Cuando tras seis u ocho semanas de tratamiento con cualquiera de estos fármacos en monoterapia no se observa efectividad, o existen importantes efectos secundarios se puede añadir un segundo inmunosupresor o sustituirlo por otro. En nuestra experiencia se intenta añadir un segundo fármaco, que tenga efectos sinérgicos con el primer tratamiento o bien pasar a emplear terapias biológicas.
Tratamiento inmunosupresor con terapias biológicas
Existen fármacos contra el factor de necrosis tumoral (anti-TNFa) introducidos en el régimen terapéutico de las AIJ. En la actualidad hay comercializados tres fármacos:
1. Infliximab (Remicade®) es un anticuerpo monoclonal quimérico contra el TNF-a que se administra por vía intravenosa en pauta de 3-5mg/Kg/8 semanas asociado a metotrexate.
2. Etanercept (Enbrel®), proteína de fusión frente al receptor soluble de la molécula del TNF-a a la que se une bloqueándola; se inyecta por vía subcutánea en pauta de 50 mg/semana.
3. Adalimumab (Humira®), anticuerpo monoclonal humanizado contra el TNF que se administra por vía subcutánea en pauta de 40 mg/2 semanas. Lehman y col38 y Biester y col39 han demostrado buena respuesta con mínimos efectos adversos utilizando este fármaco.
La mayoría de las series publicadas con estos tratamientos son series de casos abiertas, retrospectivas, en los que se incluyen pacientes con distintos tipos de uveítis. Hacen falta más ensayos multicéntricos y aleatorizados para valorar adecuadamente la eficacia de estas terapias. Estudios retrospectivos realizados por Saurenmann40, Richards41 y Rajaraman42 sobre el uso de infliximab y etanercept en uveítis en AIJ han demostrando buena respuesta clínica y una clara disminución del número de recaídas. Smith43, en estudio doble ciego y controlado con placebo en un grupo de pacientes con uveítis asociada a AIJ, no encontraron diferencias entre el grupo tratado con etanercept versus el tratado con placebo.
Existen estudios clínicos de otras terapias biológicas como anakinra (antagonista del receptor de la IL-1), atlizumab (bloqueante del receptor de la IL-6) o abatacept (inhibidor del ligando CD28 co-estimulador de las moléculas de superficie de células T) que se mantienen en el campo experimental44.
Bibliografía
1. Wakefield D, Dunlop I, McClusky PJ, Penny R. Uveitis: aetiology and disease associations in an Australian population. Aust N Z J Ophthalmol 1986; 14: 181-187. [ Links ]
2. Wakefield D, Montanaro A, McCluskey P. Acute anterior uveitis and HLA-B27. Surv Ophthalmol 1991; 36: 223-232. [ Links ]
3. Rothova A, Veenedaal WG, Linssen A, Glasius E, Kijlstra A, de Jong PT. Clinical features of acute anterior uveitis. Am J Ophthalmol 1987; 103: 137-145. [ Links ]
4. Bañares A, Jover JJ, Fernández-Gutierrez B, García J, Benítez del Castillo JM, Vargas E et al. Patterns of uveitis as a guide in making rheumatologic and immunologic diagnoses. Arthritis Rheum 1997; 40: 358-370. [ Links ]
5. Bañares A, Jover JA, Fernández B, Benítez del Castillo JM, García J, Gonzalez F et al. Bowel inflammation in anterior uveitis and spondyloarthropathy. J Rheumatol 1995; 22: 1112-1117. [ Links ]
6. Benítez del Castillo JM, García-Sánchez J, Iradier MT, Bañares A. Sulfasalazine in the prevention of anterior uveitis associated with ankylosing spondylitis. Eye 2000; 14: 340-343. [ Links ]
7. Nussenblatt RB, Whitcup SM, Palestine AG. Uveitis: Fundamentals and clinical practice. Mosby-Year Book Inc, 1996, 2nd Edition, St. Louis, USA: 265-278. [ Links ]
8. Brewerton DA, Nicholls A, Hart FD, Caffrey M, James DC, Sturrock RD. Ankylosing spondylitis and HLA-B27. Lancet 1973; 1: 904-907. [ Links ]
9. Masi AT. Epidemiology of B27 associated disease. An Rheum Dis 1979; 38: 131-134. [ Links ]
10. Keat A. Reiter´s syndrome and reactive arthritis in perspective. N England J Med 1983; 39: 1606-1615. [ Links ]
11. Lee DA, Barker SM, Su WPD, Allen GL, Liesegang TJ, Ilstrup DM. The clinical diagnosis of Reiter´s syndrome. Ophthalmology 1986; 93: 350-356. [ Links ]
12. Shivananda S, Lennard-Jones J, Logan R, Fear N, Price A, Carpenter L et al. Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease. Gut 1996; 39: 690-697. [ Links ]
13. Turkcapar N, Toruner M, Soykan I, Aydintug OT, Cetinkaya H, Duzqun N et al. The prevalence of extraintestinal manifestations and HLA association in patients with inflammatory bowel disease. Rheumatol Int 2006; 26: 663-668. [ Links ]
14. Grant IR. Zoonotic potential of Mycobacterium avium ssp paratuberculosis: the current position. J Appl Microbiol 2005; 98 (6): 1282-1293. [ Links ]
15. Das KM. Relationship of extraintestinal involvements in inflammatory bowel disease: new insights into autoimmune pathogenesis. Dig Dis Sci 1999; 44: 1-13. [ Links ]
16. Mondéjar JJ, Díaz-LLopis M. Inflamaciones Intestinales Crónicas, en Manifestaciones Oftalmológicas de las Enfermedades Generales. Sánchez Salorio M, Díaz-LLopis M, Benítez del Castillo JM, Rodríguez Ares MT. Ponencia de la Sociedad Española de Oftalmología 2001. [ Links ]
17. Ghanchi FD, Rembacken BJ. Inflammatory bowel disease and the eye. Surv Ophthalmol 2003; 48: 663-676. [ Links ]
18. Hernández-García C, Collado P, Fernández-Gutiérrez B. Manifestaciones oculares de la artritis reumatoide. Queratoconjuntivitis seca. Rev Esp Reumatol 1997; 24: 190-195. [ Links ]
19. Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase III Study Group. Ophthalmology 2000; 107: 631-639. [ Links ]
20. Cassidy JT. Artritis idiopática juvenil. In: Harris EH, Budd RC, Firestein GS, et al (Eds) Rheumatology 7th ed. Philadelphia. Elsevier Saunders; 2006; 1593-1607. [ Links ]
21. Fink CW. Proposal for the development of classification criteria for idiopathic arthritides of childhood. J Rheumatol 1995; 22: 1566-1569. [ Links ]
22. Petty RE, Southwood TR, Baum J, Bhettay E, Glass DN, Manners P et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol 1998; 25: 1991-1994. [ Links ]
23. Nocton JJ, Miller LC, Tucker LB, Schaller JG. Human parvovirus B19 associated arthritis in children. J Pediatr 1993; 122: 186-190. [ Links ]
24. Zak M, Pedersen FK. Juvenile chronic arthritis into adulthood:a long-term follow-up study. Rheumatology 2000; 39: 198-204. [ Links ]
25. Cassidy JT, Petty RE. Juvenile rheumatoid arthritis. In:Cassidy JT, Petty RE (Eds). Pediatric Rheumatology, 4th ed, Philadelphia. Saunders Company 2001; 218-225. [ Links ]
26. Sherry DD, Stein LD, Reed AM, Schanberg LE, Kredich DW. Prevention of leg length discrepancy in young children with pauciarticular juvenile rheumatoid arthritis by treatment with intraarticular steroids. Arthritis Rheum 1999; 42: 2330-2334. [ Links ]
27. Cassidy J, Kivlin J, Lindsley C, Nocton J. Ophthalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics 2006; 117: 1843-1845. [ Links ]
28. Bodhaghi B, Terrada C, Lehoang P. Cataract surgery in chilhooh uveitis. Int Ophthalmol Clin 2008; 48: 173-187. [ Links ]
29. Packham JC, Hall MA. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology 2002; 41: 1428-1435. [ Links ]
30. López Gonzalez R, Pato Cour E. Artritis idiopática Juvenil. En: Díaz Valle D, Méndez Fernández R, Benítez del Castilo JM. Actualización en el tratamiento de las uveítis. Edit. Sociedad Española de Oftalmología 2007; 97-104. [ Links ]
31. Bloch-Michel E, Nussenblat RB. International uveítis study group recommendations for the evaluation of intraocular inflamatory disease. Am J Ophthalmol 1987; 103: 234-235. [ Links ]
32. Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclatura (SUN) working group: standardization of uveitis nomenclature for reporting clinical data:results of the first international workshop. Am J Ophthalmol 2005; 140: 509-516. [ Links ]
33. Samson CM, Ekong A, Foster CS. Uveitis in children: diagnosis and management. Int Ophthalmol Clin 2001; 41: 199-216. [ Links ]
34. Woo P, Southwood TR, Prieur AM, Dore CJ, Grainger J, David J et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum 2000; 43: 1849-1857. [ Links ]
35. Kilmartin DJ, Forrester JV, Dick D. Cyclosporin A therapy in refractory non-infectious childhood uveitis. Br J Ophthalmol 1998; 82: 737-742. [ Links ]
36. Doycheva D, Deuter C, Stuebiger N, Biester S, Zierhut M. Mycophenolate mofetil in the treatment of uveitis in children. Br J Ophthalmol 2007; 91: 180-184. [ Links ]
37. Jabs DA, Rosenbaum JT, Foster CS, Holland GN, Jaffe GJ, Louie JS et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol 2000; 130: 492-513. [ Links ]
38. Vazquez-Cobian LB, Flynn T, Lehman TJ. Adalimumab therapy for childhood uveíitis. J Pediatr 2006; 149: 572-575. [ Links ]
39. Biester S, Deuter C, Michels H, Haefner R, Kuemmerle- Deschner J, Doycheva D et al. Adalimumab in the therapy of uveitis in childhood. Br J Ophthalmol 2007; 91 : 319-324. [ Links ]
40. Saurenmann RK, Levin AV, Rose JB, Parker S, Rabinovitch T, Tyrrell PN et al. Tumour necrosis factor alpha inhibitors in the treatment of childhood uveitis. Rheumatology 2006; 45: 982-989. [ Links ]
41. Richards JC, Tay-Kerney ML, Murray K, Manners P. Infliximab for juvenile idiopathic arthritis-associated uveitis. Clin Exp Ophthalmol 2005; 33: 461-468. [ Links ]
42. Rajaraman RT, Kimura Y, Li S, Haines K, Chu DS et al. Retrospective case review of paediatric patients with uveitis with infliximab. Ophthalmology 2006; 113: 308-314. [ Links ]
43. Smith JA, Thompson DJ, Whitcup SM, Suhler E, Clarke G, Smith S et al. A randomized, placebo-controlled, double-masked clinical trial of etanercept for the treatment of uveitis associated with juvenile idiopathic arthritis. Arthritis Rheum 2005; 53: 18-23. [ Links ]
44. Horneff G. Importance of the new biologicals and cytokine antagonist in the treatment of juvenile idiopathic arthritis. Z Rheumatol 2005; 64: 317-326. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
José Manuel Benítez del Castillo
Unidad de Superficie e Inflamación Ocular
Hospital Clínico San Carlos
Martín Lagos, s/n
28040 Madrid
Tfno. 913303963
Fax 913303975
E-mail: jbenitez.hcsc@salud.madrid.org

