










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.31 supl.3 Pamplona 2008
Manifestaciones corneales en las enfermedades sistémicas
Corneal manifestations in systemic diseases
J. Zarranz-Ventura, E. De Nova, J. Moreno-Montañés
Departamento de Oftalmología. Clínica Universitaria de Navarra. Pamplona.
Dirección para correspondencia
RESUMEN
Un gran número de enfermedades sistémicas presentan manifestaciones corneales dentro de su espectro de enfermedad. El estudio detallado de todos los cuadros que asocian patología corneal resulta inabarcable, por ello se presentan las enfermedades más prevalentes o características. Este estudio contempla las enfermedades pulmonares y conectivopatías (colagenosis, enfermedades reumatológicas y enfermedades inflamatorias idiopáticas), las enfermedades dermatológicas, cardiovasculares, hematológicas y la patología digestiva y hepatopancreática.
Se contemplan también, por ocasionar alteraciones corneales, las enfermedades endocrinas y metabólicas con algunas situaciones de malnutrición y estados carenciales, las infecciones sistémicas y las enfermedades renales.
Otro área que produce afectación corneal es la patología otorrinolaringológica y las enfermedades genéticas. Se repasa brevemente la toxicidad y las alteraciones corneales provocadas por fármacos.
Palabras clave: Enfermedades sistémicas. Córnea.
ABSTRACT
Systemic diseases affecting the cornea have a wide range of manifestations. The detailed study of all pathologies that cause corneal alteration is unapproachable, so we have centered our interest in the most prevalent or characteristic of them. In this paper we have divided these pathologies in sections to facilitate their study. Pulmonar and conective tissue (like colagen, rheumatologic and idiopathic inflamatory diseases), dermatologic, cardiovascular, hematologic, digestive and hepatopancreatic diseases with corneal alteration are described. Endocrine and metabolic diseases, malnutrition and carential states are also studied, as well as some otorhinolaryngologic and genetic diseases that affect the cornea. Finally, a brief report of ocular toxicity induced by drugs is referred.
Key words: Systemic diseases, cornea.
Introducción
Un gran número de enfermedades sistémicas presentan manifestaciones corneales dentro de su espectro de enfermedad. En algunos casos dicha afectación se produce en estadios avanzados y su hallazgo resulta meramente anecdótico, mientras que en otros la detección precoz de la alteración corneal puede ser determinante para alcanzar un pronto diagnóstico del cuadro sistémico e instaurar el tratamiento oportuno. Ambas situaciones justifican el estudio detallado de dichas entidades para identificarlas en nuestra práctica diaria y contribuir a definir el diagnóstico adecuado para cada paciente en particular. Se exponen los principales cuadros sistémicos con correlato patológico a nivel corneal por grupos de enfermedades, para realizar un repaso ordenado de la mayoría de ellos.
Enfermedades pulmonares
En casos de tuberculosis pulmonar la incidencia de manifestaciones oculares es del 1-2%1. En la primoinfección la afectación corneal es rara y se produce en forma de queratoconjuntivitis por inoculación directa, mientras que en reinfecciones y en las formas sistémicas puede darse de forma poco frecuente queratitis intersticial por reacción alérgica a las tuberculinas, mediada por un mecanismo de hipersensibilidad tipo IV2. Un síndrome de Sjögren secundario puede aparecer en casos de silicosis y enfermedad por polvo de grano. El síndrome de Kartagener puede producir diversas alteraciones corneales, pero a menudo se presenta sin repercusión clínica. Los broncodilatadores empleados en el asma crónico como el bromuro de ipratropio y algunos antihistamínicos producen un síndrome de ojo seco con disminución de la secreción mucoide y lagrimal, causando una intolerancia a las lentes de contacto o agravamiento de una queratoconjuntivitis seca.
Conectivopatías
Colagenosis
Un 25% de los casos de esclerodermia cursan con queratoconjuntivitis seca, con presencia de filamentos mucosos aparentes en tinción con rosa de bengala y test de Schirmer alterado. Menos frecuente es el queratocono, aunque existen casos descritos en la bibliografía3. La granulomatosis de Wegener debuta en el 16% de los casos con alteraciones oculares, siendo la manifestación más frecuente la queratitis marginal ulcerativa (hasta en un 28% según las series) que característicamente deja una zona libre entre el limbo y la zona afecta. Se ha postulado la vasculitis oclusiva necrotizante de las arterias ciliares anteriores a su paso entre los músculos rectos y la esclera como mecanismo patogénico. Con menor frecuencia se describen casos de granulomas corneales en esta entidad4.
La manifestación ocular más frecuente (88%) en el lupus eritematoso sistémico es la queratopatía epitelial puntiforme, estando descrita la queratoconjuntivitis seca por sindrome de Sjögren secundario y una queratitis periférica con adelgazamiento marginal no infiltrativo asintomático, así como nódulos límbicos flictenulares necróticos5. Puede darse una infiltacion en banda del estroma corneal por material blanco-grisáceo en algunos casos. La panarteritis nodosa cursa con un surco ulcerativo periférico que puede llegar a extenderse circularmente por todo el limbo hasta formar un anillo ulcerado. A esta afectación puede suceder una queratitis central, con vascularización, cicatrización, y hasta perforación asociada6. La queratoconjuntivitis seca (Fig. 1) es la principal manifestación ocular del síndrome de Sjögren, cursando con sequedad por destrucción de glándulas lagrimales y salivares con infiltración linfocitaria. En estadios iniciales el diagnóstico puede ser difícil ya que puede haber de forma paradójica crisis de lagrimeo antes de la completa destrucción de los acini por la infiltacion linfocitaria7. Existen 3 grados de afectación, reflejados en la tabla 1. El síndrome de Ehlers-Danlos asocia de forma característica ectasias corneales como el queratocono y el queratoglobo, llegando a alcanzar valores paquimétricos de un tercio del grosor normal. Esta fragilidad corneal motiva roturas espontáneas de la córnea ante pequeños traumatismos e hidrops agudos por roturas de la membrana de Descemet8. El subtipo VI o forma oculus fragilis puede presentar afectación corneal más severa, con embriotoxon anterior (halo corneal periférico), microcórnea o megalocórnea con glaucoma asociado, esclerocórnea periférica y córnea plana además de los cuadros antes citados9.


Enfermedades reumáticas
Dentro de las enfermedades reumatológicas, la artritis reumatoide, la artropatía psoriásica y la policondritis recidivante presentan como afectación más frecuente la queratoconjuntivis seca, a menudo como consecuencia de un síndrome de Sjögren secundario. Afecta más frecuentemente a mujeres (9:1) y suele aparecer entre la cuarta y quinta década de la vida, con irritación ocular crónica, sensación de cuerpo extraño y sequedad. Pueden llegar a producir úlceras estériles periféricas (Fig. 2), que dan lugar a adelgazamientos progresivos e indoloros que pueden evolucionar a perforación o queratitis necrotizantes10.

En su forma juvenil, la artritis reumatoide asocia en algunos casos queratopatía en banda al síndrome de ojo seco antes citado. La espondilitis anquilopoyética cursa con uveítis anteriores de repetición, que pueden provocar precipitados endoteliales (Fig. 3). Por último, el síndrome de Reiter es una artritis reactiva con combinaciones variables de lesiones urogenitales y mucocutáneas asociada al HLA-B27 que puede acompañarse de forma poco frecuente con queratitis punteadas epiteliales, infiltrados estromales y erosiones con pérdida del epitelio central de la córnea que habitualmente no dejan secuelas.

Enfermedades inflamatorias de origen desconocido
La queratitis punteada o marginal es la afectación corneal más frecuente en la enfermedad granulomatosa crónica y en la enfermedad de Kawasaki, acompañada de pannus e infiltrados perilimbares en el primer lugar y de precipitados endoteliales post-uveíticos en el segundo11. En la sarcoidosis se produce una hiposecreción lagrimal con queratitis intersticiales de repetición, con presencia de precipitados queráticos. Aunque infrecuente, puede producirse una descompensación endotelial en estas zonas, llegando a producir opacidades estromales focales.
Enfermedades dermatológicas
La dermatitis atópica cursa con queratitis punctata superficial, xeroftalmía, pannus, opacidades estromales y más raramente adelgazamiento periférico con opacificación, vascularización y queratocono12. En casos en que es preciso realizar una queratoplastia penetrante el pronóstico es peor en estos pacientes por alteración de la lágrima y de la superficie ocular asociada13. La afectación corneal más frecuente en la psoriasis es la queratitis punctata por sequedad ocular, pudiendo aparecer opacidades estromales periféricas o infiltrados subepiteliales diseminados14. En ocasiones se produce un adelgazamiento corneal periférico progresivo que puede conducir a la perforación del globo15. El tratamiento de la enfermedad sistémica con luz ultravioleta empeora el cuadro de ojo seco inicial16. Similar cuadro presenta el acné rosácea, con queratopatía punctata superficial secundaria a xeroftalmía y ocasional pérdida visual debida a complicaciones corneales. La más severa de ellas es la queratitis ulcerativa, que empieza en forma de pannus en hemicórnea inferior con opacidades subepiteliales y evoluciona a ulceración y vascularización, llegando incluso a producir perforación y endoftalmitis17.
La afectación corneal en el pénfigo es rara pero severa, pudiendo producirse laceración, ulceración e incluso perforación corneal. Más frecuente es la queratopatía por exposición del penfigoide cicatricial, con defectos epiteliales, neovascularización corneal y leucomas, con la consiguiente pérdida visual18. En ocasiones se produce la obliteración de los conductos de salida de la glándula lagrimal, causando un síndrome de ojo seco severo, y en última instancia un anquilobléfaron completo con obliteración de los fondos de saco conjuntivales y superficie ocular totalmente queratinizada y opaca19-20. Este síndrome de ojo seco severo por afectación de los conductos puede darse en otras patologías, como en la necrolisis epidérmica tóxica o síndrome de Lyell, estando descritos casos con perforación corneal y pérdida del globo ocular subsiguiente21. En el eritema multiforme y especialmente en su forma más severa, el síndrome de Stevens-Johnson, la córnea puede llegar a ulcerarse y perforarse durante el estadio de conjuntivitis severa purulenta. En estos casos pueden producirse cicatrices conjuntivales o lesiones permanentes en los anejos oculares, con síndrome de ojo seco, queratitis por exposición y neovascularizacion corneal22.
La epidermolisis bullosa cursa con pequeños quistes en las capas basales del epitelio en la periferia media de la córnea, que al migrar hacia planos más superficiales producen erosiones corneales recurrentes. Estas lesiones son debidas a la vacuolización de las células basales del epitelio corneal, causando opacidades puntiformes, difusas y granulares que alteran la visión. Además, puede aparecer triquiasis, neovascularización corneal, infiltrados granulomatosos e incluso perforación corneal en estos casos23. En la ictiosis vulgar ligada al X aparecen múltiples y finas opacidades puntiformes en el estroma corneal profundo, inmediatamente anteriores a la membrana de Descemet. Estas lesiones normalmente asintomáticas ayudan a identificar a mujeres portadoras, dado que están presentes en ellas y en pacientes con la forma ligada al X pero no en la ictiosis vulgar24.
El xeroderma pigmentosum cursa con opacidades corneales severas secundarias a queratitis por exposición con neovascularización asociada, formando leucomas que en ocasiones llegan a ulcerarse, pudiendo originar carcinomas escamosos. También se ha encontrado asociación con distrofia nodular en banda y queratocono25. En los casos que precisan queratoplastia, la microscopía de los botones corneales a los meses del injerto muestra severa desestructuración corneal a todos los niveles, degeneración de las células basales del epitelio, rotura de la membrana de Bowman (sustituida por epitelio degenerativo que se introduce en el estroma), acúmulos de colágeno en la membrana de Descemet y gránulos de melanina en células endoteliales, cuyo recuento está disminuido26. La afectación corneal en la acrodermatitis enteropática consiste en presencia de opacidades corneales subepiteliales, además de adelgazamiento, pérdida de la membrana de Bowman, neovascularización, prominencia de los nervios y cicatrización del estroma anterior27.
Enfermedades cardiovasculares
La afectación corneal en las enfermedades cardiovasculares es derivada de una hipoperfusión del segmento anterior por un defecto a nivel de los troncos supraaórticos. Así, puede darse una isquemia crónica del segmento anterior con edema corneal difuso y pliegues en endotelio en casos de insuficiencia carotídea y en la ateroesclerosis, pudiendo aparecer una queratitis neuroparalítica por ateromas en grandes vasos cerebrales28. Las fístulas carótido-cavernosas con fístula directa manifiestan dicho cuadro en un 20%, mientras que la arteritis de Takayasu puede asociar rubeosis de iris y glaucoma neovascular en casos avanzados29-30.
Enfermedades hematológicas
En el mieloma múltiple se produce un exceso de síntesis de inmunoglobulinas que aumenta los niveles plasmáticos de las mismas a títulos muy elevados, lo que causa su depósito en distintos lugares del organismo. A nivel corneal se produce el acúmulo de estos cristales en epitelio y estroma, pudiendo afectar a la visión si el depósito se da en la región central de la córnea. En estos casos puede ser precisa la realización de una queratoplastia lamelar o penetrante para recuperar la función visual del paciente. Asimismo, se ha documentado la presencia de acúmulos de células mielomatosas en el endotelio corneal y se han descrito casos de córnea verticillata en pacientes afectos de mieloma múltiple31-32. Por último, si se produce una hipercalcemia prolongada puede aparecer como secuela queratopatía en banda, haciendo necesario instaurar tratamiento con quelantes de calcio. En algunas leucemias se produce infiltración directa de las estructuras oculares por las células blásticas, produciendo infiltrados y ulceraciones corneales límbicas aparentes a la biomicroscopía con lámpara de hendidura33.
Otra entidad que cursa con afectación corneal por depósito es la macroglobulinemia de Waldenström, en la que se puede observar el depósito de cristales en estroma posterior de córnea y el agrupamiento y segmentación de la columna de eritrocitos en los vasos conjuntivales tras examen cuidadoso en lámpara de hendidura34. La gammapatía monoclonal benigna cursa con depósito de cristales en córnea, y clínicamente se manifiesta como opacidades amarillas, blanco-grisáceas o marrón-grisáceas, distribuidas de forma localizada o difusa en el espesor corneal, pudiendo ser la primera manifestación de la enfermedad35. Los pacientes trasplantados de médula ósea presentan una serie de alteraciones corneales típicas, derivadas de la toxicidad de los fármacos empleados en algunos casos como la queratoconjuntivitis por metotrexate y azatioprina o la aparición de patógenos oportunistas ante el estado de inmunosupresión del paciente, como queratitis bacterianas y herpéticas de repetición. Mención especial merece la enfermedad del injerto contra el huésped, proceso de rechazo inmunológico en el que se produce una marcada sequedad de mucosas conocida como síndrome Sjögren-like, apareciendo una severa queratoconjuntivitis seca, con xerostomía, vaginitis y xerosis cutánea acompañante36. A nivel ocular se pueden producir erosiones y ulceraciones corneales graves y recurrentes, que pueden llegar a la perforación, tanto de origen trófico como infeccioso. Habida cuenta de la severidad del cuadro, es preciso llevar a cabo un estrecho control en estos pacientes para detectar alteraciones de modo precoz e instaurar tratamiento con lubricantes e inmunosupresores en estadios iniciales, incluso presintomáticos37-38.
Enfermedades digestivas y hepatopancreáticas
Entre el 6 y el 20% de los pacientes con enfermedad de Crohn presentan algún tipo de afectación ocular, y de éstos entre un 4,5 y un 11,7 % presentan afectación corneal, en forma de infiltrados epiteliales y subepiteliales de localización periférica y yuxtalimbar, con formación de pequeñas elevaciones nodulares. La respuesta de estos infiltrados a la corticoterapia es variable, tendiendo a la cronicidad y dejando un ligero adelgazamiento parenquimatoso a la remisión de los mismos. Otras manifestaciones corneales son la aparición de opacidades intraparenquimatosas periféricas o ulceraciones paralelas al limbo con evolución tórpida y mala respuesta a tratamiento local, que desaparecen con la remisión del brote intestinal39. Por el contrario, la colitis ulcerosa presenta manifestación ocular en menos del 10% de los casos, siendo la uveítis anterior no granulomatosa el cuadro más frecuente, sin afectación corneal de interés.40 La cirrosis biliar primaria presenta queratoconjuntivitis seca en el 75% de los pacientes, pudiendo aparecer síntomas oftalmológicos propios del amplio espectro de enfermedades asociadas, como esclerodermia, artritis reumatoide, anemia perniciosa, tiroiditis, síndrome Cres41.
La afectación más característica de la degeneración hepatolenticular o enfermedad de Wilson es el anillo corneal de Kayser-Fleischer, constituido por el depósito en periferia de granulaciones de cobre muy finas de color verde-dorado, que se sitúan en la parte más posterior del estroma y en la membrana de Descemet. Cronológicamente aparece en las porciones superior e inferior de la córnea para después formar el anillo completo, cuyo grosor oscila entre 2 y 3 mm. No produce ningún tipo de alteración anatómica ni funcional de las estructuras corneales, y no es específico de esta patología, apareciendo también en la cirrosis biliar primaria y otras enfermedades que cursen con retención de cobre42. La enfermedad de Whipple rara vez presenta afectación ocular (<3%) y la afectación corneal es casi anecdótica, estando descrita la presencia de infiltrados corneales y casos de queratitis filamentosa en el contexto de un síndrome seco43. Los estados de malabsorción presentan alteraciones diversas, como opacidades corneales por déficit de zinc, queratomalacia por déficit de proteínas o distrofia corneal y xeroftalmia por déficit de vitamina A44. La cirrosis hepática y las hepatitis autoinmunes cursan con queratoconjuntivitis seca, además de asociar estados carenciales determinados en cada caso particular.
Un cuadro raro es el síndrome de Allgrove o de la triple A (Alacrimia-Acalasia-Insuficiencia adrenocortical), rara enfermedad congénita cuya manifestación más precoz y frecuente es la alacrimia, basal y refleja, que puede pasar desapercibida y ser asintomática o producir cuadros de xeroftalmía con queratitis punteada superficial e hipoestesia corneal45. El cuadro se debe a una anomalía en la inervación vegetativa del sistema lagrimal secretor, habiéndose observado en algún caso la agenesia de la glándula lagrimal46. Las manifestaciones típicas del síndrome de Alagille o displasia arterio-hepática consisten en agenesia del segmento anterior con embriotoxon y anomalía de Axenfeld, estando descritas otras anomalías como queratopatía en banda. Algunas líneas de quimioterapia empleadas en neoplasias de estómago producen daño corneal y síndrome de Sjögren secundario.
Enfermedades endocrinas
La afectación corneal más frecuente de la diabetes mellitus es la disminución de la sensibilidad corneal y la escasa respuesta cicatricial epitelial. Ambos conceptos están relacionados, dado que la hipoestesia corneal se correlaciona con el grado de sensibilidad vibratoria presente en el paciente, que a su vez ocasiona la falta de estímulo necesario para el crecimiento epitelial y la regeneración adecuada de las lesiones47-48. Este hecho asociado a la existencia de anomalías en los hemidesmosomas y en la membrana basal engrosada del epitelio corneal, favorecen la aparición de queratitis punteadas superficiales, erosiones recidivantes o úlceras estériles neurotróficas.
Cualquier tipo de depresión del sistema inmune asociada en estos pacientes facilita la sobreinfección de estas heridas por patógenos habituales y oportunistas. Asimismo, pueden producirse pliegues en la membrana de Descemet, indicando la presencia de células endoteliales anómalas49-50. La enfermedad de Graves-Basedow cursa con exoftalmos de grado variable, llegando en casos severos a producir retracciones palpebrales importantes que hacen que el cierre activo del párpado sea ineficaz provocando una continua exposición de la córnea con el desarrollo de úlceras, que pueden sobreinfectarse e incluso perforarse. Puede aparecer en estos pacientes queratoconjuntivitis límbica superior, siendo un signo de mal pronóstico para la oftalmopatía tiroidea51.
La afectación corneal más característica del hiperparatiroidismo es la queratopatía en banda que, aunque rara, es uno de los signos más frecuentes asociados con la hipercalcemia secundaria a la alteración hormonal de estos pacientes52. Se produce por el depósito intracelular de cristales de hidroxiapatita cálcica en las células basales del epitelio de la córnea, membrana de Bowman y estroma anterior, provocando opacidades blancogrisáceas en área interpalpebral a lo largo del meridiano horizontal de la córnea (Fig. 4). El epitelio que descansa sobre la queratopatía permanece intacto en la mayoría de los casos. Inicialmente los depósitos comienzan en periferia separados del limbo por una estrecha banda de córnea normal, y progresan centrípetamente de forma gradual, dejando multitud de pequeños agujeros relacionados con la entrada de nervios corneales no calcificados, hasta que ocupan la porción central de la córnea afectando a la visión de forma severa. Esta entidad no es exclusiva de este cuadro, y puede aparecer en diversas enfermedades, algunas de las cuales están recogidas en la tabla 2.


En el hipoparatiroidismo y pseudohipoparatiroidismo se produce una queratoconjuntivitis con inyección conjuntival, queratopatía superficial con vascularización marginal y fotofobia, relacionadas con el descenso de secreción lagrimal y en el contexto de un proceso autoinmune53. El síndrome de neoplasia endocrina múltiple (MEN) tipo 2B asocia de forma característica la existencia de prominentes nervios corneales presentes desde temprana edad en el 100% de los pacientes54. Histológicamente estos nervios corresponden a numerosos axones desmielinizados y a multitud de células de Schwann, que los hace visibles a la biomicroscopía con lámpara de hendidura55. Se ha descrito esta alteración en el MEN subtipo 2A, por lo que varios autores sugieren un posible origen genético común entre ambas entidades, sitas en el cromosoma 1056-57. Ambos síndromes cursan con queratoconjuntivitis seca (hasta en el 67% en el subtipo 2B) asociando en menor frecuencia la presencia de vasos prominentes alrededor del limbo.
Enfermedades metabólicas
La amiloidosis produce afectación corneal variable en sus distintas formas, localizadas y generalizadas. En las amiloidosis localizadas se producen distintos cuadros englobados en el término amiloidosis corneal primaria, como las distrofias reticulares tipo I de Biber-Haab-Dimmer o los tipos II y III. La afectación corneal en las amiloidosis sistémicas es rara y varía en función del subtipo. En la amiloidosis heredofamiliar tipo I se produce xeroftalmía por alteración de la glándula lagrimal, asociando una queratitis neuroparalítica con úlceras, neovascularización e incluso perforaciones de la córnea mientras que en la forma heredofamiliar tipo IV puede aparecer distrofia reticular de tipo II, comenzando a aparecer en la tercera década de la vida58. La hemocromatosis cursa con depósitos de hierro próximos al limbo y asocia frecuentemente un síndrome de ojo seco, además de córnea pseudoverticillata59. En la gota se produce la llamada distrofia úrica de la córnea, que consiste en la presencia de opacidades amarillentas subepiteliales próximas al limbo con extensión central que pueden acompañarse de úlceras y neovascularización asociada. Este cuadro poco frecuente se origina por depósitos de cristales de urato monosódico y al igual que las porfirias puede cursar con queratopatía en banda.
Alteraciones en el metabolismo de aminoácidos
En la cistinosis se produce el acúmulo intralisosomal de cristales de cistina, siendo la alteración corneal la más típica de las manifestaciones oculares, con una incidencia del 90% en niños menores de un año. Los cristales se localizan tanto en la región central como periférica de la córnea y se acumulan en el epitelio para después progresar hacia el endotelio, afectando a todo el espesor corneal. Son frecuentes las úlceras de repetición, que llegan a inducir neovascularización asociada. En casos avanzados esta indicada la realización de una queratoplastia penetrante, a pesar de que el injerto será invadido por los cristales60. En la homocistinemia aparece queratitis en el 3% de los casos y en la ocronosis o alcaptonuria se puede producir pigmentación del estroma paralímbico, aunque de forma poco frecuente. La tirosinosis cursa con episodios de queratoconjuntivitis bilateral, recidivante y pseudodendrítica que en ocasiones pueden asociar úlceras geográficas con edema corneal (Fig. 5) y neovascularización asociada que evolucionan hacia opacidades corneales61.

Alteraciones en el metabolismo de carbohidratos
En la glucogenosis tipo I subtipo enfermedad de Von Gierke por déficit de glucosa-6-fosfatasa se producen depósitos de colágeno en el epitelio y estroma, formando opacidades corneales periféricas nubosas y delimitadas. Similar afectación se da en las mucopolisacaridosis de Hurler, Hunter, Morquio, Sanfilippo, Sheie, Maroteaux-Lamy o Sly, en las que se producen depósitos de distintos hidratos de carbono en función del nivel metabólico al que actúe la enzima deficitaria. La frecuencia de afectación varía desde el 80% del síndrome de Hurler a la muy rara afectación en el síndrome de Sanfilippo. Otras enfermedades de este grupo con cuadro similar son la fucosidosis o enfermedad de Durand, manosidosis, mucolipidosis, lipomucopolisacaridosis y galactosialidosis o síndrome de Goldberg.
Alteraciones en el metabolismo de lípidos
Las hiperlipoproteinemias (sobre todo la tipo II y en menor frecuencia III, IV y V) asocian arco senil o gerontoxon en sujetos jóvenes62. Constituido por depósitos de colesterol y fosfolípidos entre las membranas de Bowman y Descemet, consiste en una opacidad periférica blanco-grisácea arciforme, separada del limbo por una estrecha banda de córnea clara que disminuye a medida que evoluciona en tres fases (Tabla 3). Suele aparecer hacia los 60 años bilateralmente de forma fisiológica, momento en el que se denomina arco senil y no tiene relación alguna con las dislipemias. Su presencia en gente joven aunque no es patognomónica se relaciona con hipercolesterolemia, y aparece en el 47% de pacientes con hiperlipoproteinemia, aumentando su incidencia si se asocia a diabetes, HTA o tabaquismo. Su presencia en menores de 50 años es un índice de riesgo cardiovascular importante, dado que suele coexistir con ateromas coronarios, angor y antecedentes de infarto de miocardio63. En estos casos se debe hacer diagnóstico diferencial con la distrofia cristalina de Schnyder, de transmisión autosómica dominante, que se manifiesta en la segunda década de la vida y además del arco senil periférico cursa con finos cristales de fosfolípidos y colesterol en el tercio anterior del estroma corneal asociados a opacidad central gris que provoca deslumbramiento, sin otras alteraciones sistémicas.

Dentro de las hipolipoproteinemias la enfermedad de Tangier puede presentar opacidades corneales finas y difusas en el estroma profundo de la córnea central hasta en el 50% de los pacientes, aumentando con la edad y siendo casi constantes a los 50 años. La enfermedad de los ojos de pez cursa con opacidades corneales finas blanco-grisáceas dispuestas en mosaico que por su extensión e intensidad recuerdan a los ojos de un pez hervido. Debuta en la segunda década de la vida y progresa con deterioro gradual de la agudeza visual, produciéndose las opacidades por depósito de colesterol en estroma y membrana de Bowman, con respeto característico del epitelio64. Las esfingolipidosis como la enfermedad de Gaucher, la cerebrosidosis y la enfermedad de Fabry cursan con depósitos corneales en bigotes de gato o córnea verticillata (Fig. 6), mientras que las esfingomielinosis como la enfermedad de Niemann-Pick tipo A o las gangliosidosis como la enfermedad de Norman-Landing presentan opacidades corneales por depósito de distintos metabolitos.

Malnutrición y estados carenciales
Las hipovitaminosis pueden producir distintos tipos de afectación corneal. Tras la nictalopia o ceguera crepuscular por disminución de síntesis de rodopsina en los bastones, la manifestación más grave de la hipovitaminosis A es la xeroftalmía. Se produce xerosis corneal en forma de queratitis punctata y xerosis conjuntival con presencia de manchas de Bitot, pudiendo aparecer opacidades superficiales queratinizadas con neovasos que llegan desde el limbo y úlceras superficiales con edema estromal subyacente65. En casos graves puede llegarse a la queratomalacia, forma en que se produce de forma aguda una necrosis de la córnea seca y queratinizada. La infiltración masiva de polimorfonucleares en el estroma corneal hace que estos liberen enzimas colagenolíticas, ablandando la córnea y produciendo el denominado melting corneal que en determinados casos puede evolucionar hacia la ulceración y perforación de la córnea66. En estos casos el iris puede obliterar las perforaciones provocando glaucoma secundario y estafiloma corneal, agravando el cuadro. Este proceso ocurre de forma brusca en pacientes con malnutrición severa y presencia de enfermedad infecciosa aguda concurrente, pudiendo causar ceguera total si la afectación es bilateral. La queratomalacia puede considerarse un importante marcador de mortalidad, dado que en niños no tratados ésta oscila entre un 50 y 90%67. El déficit de vitamina B2 (riboflavina) puede cursar con queratitis punctata superficial y neovascularización periférica del estroma corneal, mientras que el déficit de B3 (niacina) lo hace con erosiones epiteliales e infiltración estromal. Por el contrario, las hipervitaminosis también pueden provocar alteraciones corneales, como la hipervitaminosis D (colecalciferol) que puede cursar con calcificaciones en córnea en forma de queratopatía en banda.
Enfermedades infecciosas
La tuberculosis sistémica afecta a la córnea en forma de queratitis intersticial por el mecanismo de hipersensibilidad tipo IV a las tuberculinas antes citado, produciendo frecuentemente afectación de modo unilateral con presencia de infiltrados en la capa media y profunda de la córnea. En la lepra se producen queratitis avasculares en sector temporal superior, con presencia de lepromas corneales patentes a la exploración con lámpara de hendidura como esférulas blanquecinas en el estroma. Está descrito el engrosamiento de los nervios corneales en las formas tuberculoides, asociado muy frecuentemente con hipoestesia corneal y conjuntival. La enfermedad de Lyme producida por Borrelia burgdorferi puede cursar con queratoconjuntivitis y úlceras corneales por exposición, como complicación de la parálisis facial frecuente en estos pacientes. Puede producirse también queratitis estromal con edema corneal asociado o precipitados queráticos finos, con buena respuesta a corticoides, pero recidivas frecuentes si suspensión brusca.
La manifestación corneal más característica de la sífilis es la queratitis intersticial, siendo más frecuente en las formas congénitas que en las adquiridas, donde suele ser unilateral y con menor vascularización. A diferencia de la queratitis intersticial tuberculosa, en estos casos típicamente se da respeto epitelial y endotelial, con invasión del estroma corneal por vasos profundos desde la conjuntiva límbica con infiltración celular asociada, produciendo la característica placa salmon. Tras varios meses de evolución la córnea se aclara y los vasos quedan exangües, sin perfusión, son los llamados vasos fantasma68. La infección por clamidias puede presentar distintos cuadros. En primer lugar produce el tracoma, que supone la causa más importante de ceguera prevenible a nivel mundial y consiste en una conjuntivitis folicular crónica que conduce a cicatrización de la conjuntiva y de la córnea con neovascularización asociada. La afectación corneal en estadios iniciales varía con queratitis punteada subepitelial entre leve y moderada y presencia de infiltrados periféricos epiteliales o subepiteliales, para evolucionar hacia un cuadro de conjuntivalización corneal en estadios avanzados.
Por otro lado, la infección por clamidias puede desencadenar un síndrome de Reiter, antes citado. La tularemia puede producir infección corneal en raras ocasiones, con presencia de infiltrados corneales periféricos y edema corneal agudo, mientras que en la difteria puede producirse ulceración y necrosis corneal. Las endoftalmitis bacterianas endógenas cursan con edema corneal. El virus del Herpes simple produce afectación corneal casi siempre de forma primaria. El virus varicela-zóster puede producir afectación ocular en forma de queratoconjuntivitis con adenopatías, queratitis punteada o dendrítica y lesiones numulares que pueden permanecer muchos meses, además de hipoestesia corneal asociada. El virus de Epstein-Barr cursa con queratitis estromales e infiltrados subepiteliales similares a los que aparecen en las queratitis por adenovirus, mientras que el virus de la inmunodeficiencia humana (VIH) produce un síndrome de ojo seco. En pacientes VIH positivos con queratitis epitelial punteada refractaria a tratamiento se debe descartar la queratitis por microsporidios, casi exclusiva de SIDA y por lo tanto indicador de progresión de la enfermedad. En pacientes afectos de candidiasis sistémica pueden aparecer úlceras corneales si coexiste patología corneal predisponente y tratamiento prolongado con corticoides, por lo que deben tenerse en cuenta ambos factores.
La afectación corneal por amebas difiere según el mecanismo de infección. Si esta se produce por vía digestiva, en forma parasitaria, pueden aparecer manifestaciones oculares como úlceras corneales tórpidas y recidivantes, aunque son raras. Como amebas libres pueden producir queratitis en individuos sanos, sin afectación sistémica, tipicamente asociadas a uso de lentes de contacto, traumatismo corneal o exposición a polvo o agua contaminada. La queratitis en su estadio inicial cursa con afectación epitelial de patrón dendrítico que independientemente del tratamiento puede desaparecer y recidivar, por lo que en ocasiones este cuadro se puede confundir con una queratitis por herpes simple o micótica. Más adelante aparecen infiltrados estromales densos únicos o múltiples y en anillo, que provocan la destrucción del epitelio corneal y del estroma, pudiendo evolucionar en estadios finales a descemetocele y perforación corneal. La perineuritis de los nervios corneales es muy característica de esta entidad, pero se da de forma poco frecuente. La tripanosomiasis y la leishmaniasis pueden cursar con queratitis intersticial, perforación corneal y panoftalmía, mientras que en la malaria la invasión corneal directa no ha sido descrita, pudiendo existir queratitis por exposición en la malaria cerebral. La oncocercosis o ceguera de los ríos es una de las cinco causas de ceguera prevenibles más importantes por la OMS, produciéndose manifestaciones oculares en el 50-90% en áreas endémicas. Puede aparecer queratitis punteada con opacidades lineales en relación con la migración intracorneal de las microfilarias, para luego evolucionar a queratitis esclerosante con disminución severa de la agudeza visual.
Enfermedades renales
En la insuficiencia renal crónica se produce un hiperparatiroidismo secundario que altera el metabolismo fosfocálcico, provocando un aumento de los niveles de calcio sérico. A consecuencia de ello pueden aparecer depósitos cálcicos (fosfato cálcico y apatita) en córnea y conjuntiva produciendo inflamación, ojo rojo e incluso queratopatía en banda.69 Este cuadro puede presentarse también en pacientes sometidos a tratamiento con hemodiálisis. En el trasplante renal pueden darse complicaciones derivadas de la inmunosupresión inherente al trasplante como queratitis infecciosas por herpes simple.
La afectación corneal en el síndrome de Alport es variable, pudiendo aparecer vesículas endoteliales compatibles con distrofia polimorfa posterior, opacidades subepiteliales, arco senil, microcórnea y erosiones epiteliales recurrentes70. El síndrome WARG (Tumor de Wilms–Aniridia-Retraso mental-Malformaciones genitourinarias) es un cuadro raro que puede cursar con opacidades corneales.
Enfermedades otorrinolaringológicas
El síndrome de Cogan asocia sordera neurosensorial progresiva, acúfenos y síntomas vestibulares con queratitis intersticial bilateral no supurativa con infiltrado celular y vascularización del estroma corneal sin afectación epitelial o endotelial71. La queratitis intersticial es dolorosa y suele acompañarse de inyección mixta, de iritis leve y edema palpebral. La forma inicial de presentación son unas opacidades numulares bilaterales, periféricas y subepiteliales que suelen responder bien a corticoides tópicos, pudiendo aparecer en fases tardías opacificación corneal progresiva de patrón dendritiforme alrededor de vasos neoformados en el estroma profundo con engrosamiento de la membrana de Descemet, formación de tejido conectivo y depósito de cristales siguiendo el curso de los vasos sanguíneos72.
Enfermedades genéticas
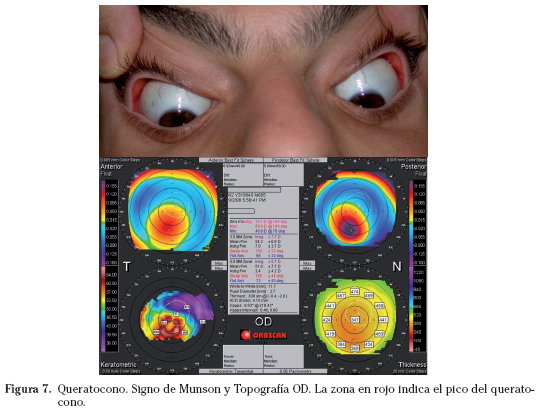
La cromosomopatía más frecuente es el síndrome de Down que cursa con queratocono en el 10% de los pacientes, pudiendo evolucionar en más de un 15% de una manera aguda.73 La afectación oscila desde queratoconos iniciales sólo detectables por estudios topográficos corneales hasta casos avanzados con un manifiesto signo de Munson (Fig. 7). La causa de una mayor incidencia de queratocono es desconocida, aunque algunos investigadores han sugerido como origen el frotamiento ocular, aunque no debe excluirse la posibilidad de alteraciones moleculares en la composición corneal. La trisomía 18 o síndrome de Edwards presenta un engrosamiento irregular de la membrana de Descemet con múltiples pliegues en el endotelio, mientras que el Síndrome de Patau (Trisomía 13) y la trisomía 8 presentan opacidades corneales como alteración más característica74-75. Por último, el síndrome de Klinefelter puede presentar de forma infrecuente queratocono posterior.
Afectación corneal por fármacos y drogas
Un amplio grupo de fármacos pueden producir alteraciones corneales. La más frecuente de ellas es la queratitis, que puede ser provocada por los conservantes de los colirios o las soluciones de limpieza de lentes de contacto (p. ej. cloruro de benzalconio, clorhexidina, timerosal) e incluso por anestésicos tópicos, corticoides, antivíricos como el aciclovir y antibióticos como la tobramicina administrados de forma tópica. La tabla 4 recoge las principales alteraciones corneales de fármacos de uso común76.

Bibliografía
1. Bodaghi B, Le Hoang P. Ocular tuberculosis. Curr Opin Ophthalmol 2000; 11: 443-448. [ Links ]
2. Helm EJ, Holland GN. Ocular tuberculosis. Surv Ophthalmol 1993; 38: 229-256. [ Links ]
3. Foster CS. Systemic lupus erythematosus, discoid lupus erythematosus and progressive systemic sclerosis. Int Ophthalmol Clin 1997; 37: 93-110. [ Links ]
4. Harman LE, Margo CE. Wegener´s granulomatosis. Surv Ophthalmol 1998; 42: 458-480. [ Links ]
5. Gloor P, Kim M, McNiff JM, Wolfley D. Discoid Lupus erythematosus presenting as asymmetric posterior blepharitis. Am J Ophthalmol 1997; 124: 707-709. [ Links ]
6. Akova YA, Jabbur NS, Foster CS. Ocular presentation of polyarteritis nodosa. Ophthalmology 1993; 100: 1775-1781. [ Links ]
7. Hikichi T, Yoshida A, Tsubota K. Lymphocytic infiltration of the conjunctiva and the salivary gland in Sjögren´s syndrome. Arch Ophthalmol 1993; 111: 21-22. [ Links ]
8. McDermott ML, Holladay J, Liu D, Puklin JE, Shin DH, Cowden JW. Corneal topography in Ehlers-Danlos syndrome. J Cataract Refract Surg 1998; 24: 1212-1215. [ Links ]
9. Cameron JA. Corneal abnormalities in Ehlers-Danlos Syndrome type VI. Cornea 1993; 12: 54-59. [ Links ]
10. Harper SL, Foster CS. The ocular manifestations of rheumatoid disease. Int Ophthalmol Clin 1998, 38: 1-19. [ Links ]
11. Ohno S, Miyajima T, Higuchi M, Yoshida A, Matsuda H, Saheki Y et al. Ocular manifestations of Kawasaki disease (mucocutaneous lymph node syndrome). Am J Ophthalmol 1982; 93: 713-717. [ Links ]
12. Dogru M, Nakagawa N, Tetsumoto K, Katakami C, Yamamoto M. Ocular surface disease in atopic dermatitis. Jpn J Ophthalmol 1999; 43: 53-57. [ Links ]
13. Reinhard T, Moller M, Sundmacher R. Penetrating keratoplasty in patients with atopic dermatitis with and without systemic cyclosporin A. Cornea 1999; 18: 645-651. [ Links ]
14. Catsarou-Catsari A, Katsambas A, Theodoropoulos P, Stratigos J. Ophthalmological manifestations in patients with psoriasis. Acta Derm Venereol 1984; 64: 557-559. [ Links ]
15. Varma S, Woboso AF, Lane C, Holt PJ. The peripheral corneal melting syndrome and psoriasis: coincidence or association? Br J Dermatol 1999; 141: 344-346. [ Links ]
16. Linden KG, Weinstein GD. Psoriasis: current perspectives with an emphasis on treatment. Am J Med 1999; 107: 595-605. [ Links ]
17. Akpek EK, Merchant A, Pinar V, Foster CS. Ocular Rosacea: Patient characteristics and follow up. Ophthalmology 1997; 104: 1863-1867. [ Links ]
18. Smith RJ, Manche EE, Mondino BJ. Ocular cicatricial pemphigoid and ocular manifestations of pemphigus vulgaris. Int Ophthalmol Clin 1997; 37: 63-75. [ Links ]
19. Kaye SB, Morton CE, Needham A, Scott JA, Watson A, Hiscott P. Superior tarsal conjunctival bullae in cicatricial pemphigoid. Am J Ophthalmol 1997; 123: 405-407. [ Links ]
20. Donnenfeld ED, Perry HD, Wallerstein A, Caronia RM, Kanellopoulos AJ, Sforza PD et al. Subconjunctival mitomycin C for the treatment of ocular cicatricial pemphigoid. Ophthalmology 1999; 106: 72-78. [ Links ]
21. Binaghi M, Koso M, Saiag P, Roujeau JC, Coscas G. Ocular involvement in Lyell´s syndrome. Incidence, evolution, prognosis. Ophthalmologie 1988; 2: 121-122. [ Links ]
22. Power WJ, Ghoraisi M, Merayo-Lloves J, Neves RA, Foster CS. Analysis of the acute ophthalmic manifestations of the erythema multiforme/Stevens-Johnson syndrome/toxic epidermal necrolysis disease spectrum. Ophthalmol 1995; 102: 1669-1676. [ Links ]
23. Tong L, Hodgkins PR, Denyer J, Brosnahan D, Harper J, Russell-Eggit I et al. The eye in epidermolysis bullosa. Br J Ophthalmol 1999; 83: 323-326. [ Links ]
24. Kempster RC, Hirst LW, de la Cruz Z, Green WR. Clinicopathologic study of the cornea in X-linked ictiosis. Arch Ophthalmol 1997; 115: 409-415. [ Links ]
25. Goyal JL, Rao VA, Srinivasan R, Agrawal K. Oculocutaneous manifestations in xeroderma pigmentosa. Br J Ophthalmol 1994; 78: 295-297. [ Links ]
26. Jalali S, Boghani S, Vemuganti GK, Ratnakar KS, Rao GN. Penetrating keratoplasty in xeroderma pigmentosum. Case reports and review of the literature. Cornea 1994; 13: 527-533. [ Links ]
27. Cameron JD, McClain CJ. Ocular histopathology of acrodermatitis enteropathica. Br J Ophthalmol 1986; 70: 662-667. [ Links ]
28. Sánchez Salorio M, Pazos González B. Enfermedades cardiovasculares. En: Sánchez Salorio M, Díaz-Llopis M, Benítez del Castillo JM, Rodríguez Ares MT. Manifestaciones oftalmológicas de las enfermedades generales. 77ª Ponencia oficial de la S.E.O. 2001; 1: 35. [ Links ]
29. Lewis JR, Glaser JS, Schatz NJ. Pulseless (Takayasu) disease with ophthalmic manifestations. J Clin Neuroophthalmol 1993; 13: 242-249. [ Links ]
30. Durcan FJ. Carotid cavernous fistula. En: Gold DH, Weingeist TA. Color atlas of the eye in the systemic disease. Lippincot. Philadelphia 2001; 627-629. [ Links ]
31. Baker TR, Spencer WH. Ocular findings in multiple myeloma: A report of two cases. Arch Ophthalmol 1974; 91: 110. [ Links ]
32. Chong EM, Campbell RJ, Bourne WM. Vortex keratopathy in a patient with multiple myeloma. Cornea 1997; 16: 592-594. [ Links ]
33. Mclean H, Clarke MP, Strong NP, Kernahan J, Ashrat S. Primary ocular relapse in acute lymphoblastic leukemia. Eye 1996; 10: 719-722. [ Links ]
34. Orellana J, Friedman AH. Ocular manifestations of multiple myeloma, Waldenströms macroglobulinemia and benign monoclonal gammapathy. Survey Ophthalmol 1981; 26: 157-169. [ Links ]
35. Hutchinson K, Del Pra M, Apel A. Immunoglobulin G crystalline keratopathy associated with monoclonal gammapathy. Aust N Z Ophthalmol 1998; 26: 177-179. [ Links ]
36. Mencucci R, Rossi Ferrini C, Bosi A, Volpe R, Guidi S, Salvi G. Ophthalmological aspects in allogenic bone marrow transplantation: Sjögren-Like syndrome in graft-versus-host disease. Eur J Ophthalmol 1997; 7: 13-18. [ Links ]
37. Ng JS, Lam DS, Li CK, Chik KW, Cheng GP, Yuen PM et al. Ocular complications of pediatric bone marrow transplantation. Ophthalmology 1999; 106: 160-164. [ Links ]
38. Kiang E, Tesavibul N, Yee R, Kellaway J, Przepiorka D. The use of topical cyclosporine. A in ocular graft-versus-host disease. Bone Marrow Transplant 1998; 22: 147-151. [ Links ]
39. Salmon JF, Wright JP, Murray AD. Ocular inflammation in Crohn´s disease. Ophthalmology 1991; 98: 480-484. [ Links ]
40. Lyons JL, Rosenbaum JT. Uveitis associated with inflammatory bowel disease compared with uveitis associated with spondyloarthropathy. Arch Ophthalmol 1997; 115: 61-64. [ Links ]
41. Tsianos EV, Hoofnagle JH, Fox PC, Alspaugh M, Jones EA, Schafer DF et al. Sjögren´s syndrome in patients with primary biliary cirrhosis. Hepatology 1990; 11: 730-734. [ Links ]
42. Wiznia RA. Wilson´s disease. En: Gold DH,Weingeist TA. The eye in the systemic disease. Lippincott JB. Philadelphia. 1990; 132: 390-392. [ Links ]
43. Williams JG, Edward DP, Tessler HH, Persing DH, Mitchell PS, Goldstein DA. Ocular manifestations of Whipple´s disease: an atypical presentation. Arch Ophthalmol 1998; 116: 1232-1234. [ Links ]
44. Watson NJ, Hutchinson CH, Atta HR. Vitamin A deficiency and xerophthalmia in the United Kingdom. BMJ 1995; 22: 1050-1051. [ Links ]
45. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet 1978; 1: 1284-1286. [ Links ]
46. Chu ML, Berlin D, Axelrod FB. Allgrove syndrome: documenting cholinergic dysfunction by autonomic tests. J Pediatr 1996; 129: 156-159. [ Links ]
47. Nielsen NV. Corneal sensitivity and vibratory perception in diabetes mellitus. Acta Ophthalmol 1978; 56: 406-411. [ Links ]
48. Nielsen NV, Lund FS. Diabetic polyneuropathy: corneal sensitivity, viratory perception and Achilles tendon réflex in diabetes. Acta Neurol Scandinav 1979; 59: 15-22. [ Links ]
49. Schultz RO, Matsuda M, Yee RW, Edelhauser HF, Schultz KJ. Corneal endothelial changes in type I and type II diabetes mellitus. Am J Ophthalmol 1984; 98: 401-410. [ Links ]
50. Olsen T, Busted N, Schmitz O. Corneal thickness in diabetes mellitus. Lancet 1980; 1: 883. [ Links ]
51. Kadrmas DF, Bartley GB. Superior límbic keratoconjunctivitis. A prognostic sign for severe Graves ophthalmopathy. Ophthalmology 1995; 102: 1472-1475. [ Links ]
52. Willis R. Endocrine disease and cornea. En Krachmer JH, Mannis MJ, Holland EJ. Cornea and external disease: clinical diagnosis and management. St Louis: Mosby; 1997: 979-988. [ Links ]
53. Stieglitz LN, Kind HP, Kazdan JJ, Fraser D, Kooh SW. Keratitis with hypoparathyroidism. Am J Ophtalmol 1977; 84: 467-472. [ Links ]
54. Fink A, Lapidot M, Spierer A. Ocular manifestations in multiple endocrine neoplasia type 2b. Am J Ophthalmol 1998; 126: 305-307. [ Links ]
55. Riley FC Jr, Robertson DM. Ocular histopathology in multiple endocrine neoplasia type 2B. Am J Ophthalmol 1981; 91: 57-64. [ Links ]
56. Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. Am J Ophthalmol 1991; 111: 307-311. [ Links ]
57. Takai S, Kinoshita S, Tanaka F, Ikeda M, Tanaka N, Kobayashi T. Prominent corneal nerves in patients with multiple endocrine neoplasia type 2A: Diagnostic implications: World J Surg 1992; 16: 620-624. [ Links ]
58. Abenhaim A, Hoang-Xuan T, Goichot-Bonnat L, Dhermy P, Pouliquen Y. Un cas de perforation cornéene spontanée au cours de l´amylose portugaise de De Andrade. Bull Soc Ophthalmol Fr 1988; 88: 225-226. [ Links ]
59. Hoissen H, Kopstad G, Elsas T, Ringvold A, Tvedt KE, Odegaard A. Idiopatic haemochromatosis and eye symptoms. A case report. Acta Ophthalmol 1985; 63: 192-197. [ Links ]
60. Marrakchi S, Lasram L, Bouguila H, Barbirou I, Ouertani A, Ayed S. Apport de lexamen ophthalmologique au diagnostique des nephropaties familiales. À propos de 10 cas. J Fr Ophtalmol 1994 ; 17: 238-241. [ Links ]
61. Godde-Joly D, Larregue M, Roussar B, Van Efenterre G. Un cas de syndrome de Richner-Hanhart. Tyrosine a manifestations oculaires, cutanées et mentales. J Fr Ophtalmol 1979; 2: 23-28. [ Links ]
62. Kanski J. Cornea. En: Kanski J. Oftalmología Clínica 5ª Edición, Madrid, Elsevier. 2006; 5: 122-123. [ Links ]
63. Rouffy P, Schilovitz G, Bakir R, Chanu B, Loeper JG, Jablon L. Contribution a l´etude des anomalies lipidiques et lipoproteiniques associés au gerontoxon. Bull Soc Ophthalmol Fr 1981; 81: 171-174. [ Links ]
64. Carlson LA. Fish-eye disease: a new familiar condition with massive corneal opacities and dyslipoproteinemia. Eur J Clin Invest 1982; 12: 41-53. [ Links ]
65. Sommer A. Nutritional factors in corneal xerophthalmia and keratomalacia. Arch Ophthalmol 1982; 100: 399-403. [ Links ]
66. Sommer A. Effects of vitamin A deficiency on the ocular surface. Ophthalmology 1983; 90: 592-600. [ Links ]
67. Sommer A, Sugana T. Corneal xerophthalmia and keratomalacia. Arch Ophthalmol 1982; 100: 404-411. [ Links ]
68. Kanski J. Cornea. En: Kanski J. Oftalmología Clínica 5ª Edición, Madrid, Elsevier. 2006; 5: 108. [ Links ]
69. Demco TA, Mc Cormick AQ, Richards JS. Conjunctival changes in chronic renal failure. Can J Ophthalmol 1974, 9: 208-213. [ Links ]
70. Colville D, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet 1997; 18: 161-173. [ Links ]
71. Robin JB, Duguel R. Immunologic disorders of the cornea and conjunctiva. En: Kauffmann HE. New York, Churchill Livingstone 1988; 511-561. [ Links ]
72. Carreras B, Guerrero JC. Inflamaciones de la córnea. En: Alió J, Carreras B, Ruiz-Moreno JM. Inflamaciones oculares. 71ª Ponencia de la S.E.O. Barcelona, Edika-Med 1995; 187-211. [ Links ]
73. Roizen NJ, Mets MB, Blondis TA. Ophthalmic disorders in children with Down syndrome. Dev Med Child Neurol 1994; 36: 594-600. [ Links ]
74. Hoepner J, Yanoff M. Ocular anomalies in trisomy 13-15. Am J Ophthalmol 1972; 74: 729. [ Links ]
75. Stark DJ, Gilmore DW, Vance JC, Perarne J. A corneal abnormality associated with trisomy 8 mosaicism syndrome. Br J Ophthalmol 1987; 71: 29-31. [ Links ]
76. Díaz-Llopis M, Benítez del Castillo JM, Belda J, Morillas PJ. Toxicidad ocular por fármacos y drogas. En: Sánchez Salorio M, Díaz-Llopis M, Benítez del Castillo JM, Rodríguez Ares MT. Manifestaciones oftalmológicas de las enfermedades generales. 77ª Ponencia oficial de la S.E.O. 2001; 19: 509-518. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
J. Zarranz-Ventura
Dpto. de Oftalmología
Clínica Universitaria
Avda. Pío XII, 36
31008 Pamplona
Tfno. 948 29 63 31 - Ext. 4242
Email: jzarranz@unav.es

