










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
En diciembre de 2019, en Wuhan (China) surgió un brote de neumonía ocasionado por un nuevo coronavirus, inicialmente llamado por la Organización Mundial de la Salud (OMS) 2019-novel coronavirus (2019-nCoV). Posteriormente, pasó a conocerse como coronavirus de tipo 2 causante del síndrome respiratorio agudo severo (SARS-CoV-2), productor de la enfermedad por coronavirus 2019 (COVID-19)1. Actualmente, está considerada un problema de salud pública global, por su rápida diseminación y sus implicaciones económicas, políticas y socio-culturales. Por este motivo, y con más de 90.000.000 de casos y 1.900.000 muertes en 191 países, fue declarada pandemia por la OMS en marzo de 20202.
Aunque se ha descrito el tropismo renal del SARS-CoV-23, su papel en la instauración de la lesión renal aguda (LRA) o el impacto en la enfermedad renal crónica (ERC), especialmente en pacientes de edad avanzada, con otras comorbilidades o inmunosupresión subyacente4, no está completamente dilucidado5,6.
Por ello, el objetivo de esta revisión es proporcionar una visión global sobre el rol del SARS-CoV-2 en la enfermedad renal aguda y crónica, y sus posibles mecanismos patogénicos en la afectación renal.
SARS-CoV-2: un nuevo miembro de la familia Coronaviridae
Origen y estructura del genoma
Los primeros casos de COVID-19 fueron detectados en personas expuestas a un mercado de mariscos de Wuhan, provincia de Hubei, China, con síntomas de neumonía atípica7. Estudios posteriores sugirieron que los murciélagos (Rhinolophus affinis) podían ser el reservorio del SARS-CoV-2, ya que la secuenciación completa del genoma arrojó una similitud del 96% con los genomas del Bat-CoV y Bat-CoV RaTG138,9. Además, los resultados de alineación de secuencias de proteínas y los análisis filogenéticos han planteado la posibilidad de hospedadores intermediarios alternativos como tortugas y pangolines10,11.
El SARS-CoV-2 pertenece a la familia Coronaviridae y al género betacoronavirus, y tiene una similitud con el SARS-CoV y el MERS-CoV de hasta un 79% y 50%, respectivamente12,13. Estructuralmente, es un virus esférico, de ARN monocatenario, con una longitud que varía entre 26-32 kbs y un diámetro de 50-200 nm8. La transcripción de proteínas no estructurales, responsables del mantenimiento del genoma y la replicación viral, depende de los dos tercios próximos al extremo 5' terminal del genoma del virus, que contienen dos marcos de lectura abiertos, ORF1 y ORF2. Las proteínas estructurales situadas en la membrana del virus, como la proteína de membrana (proteína M), de envoltura (proteína E), la nucleocápside (proteína N) y la proteína espiga (proteína S) se codifican en los marcos de lectura del tercio próximo al extremo 3' 14.
La proteína S, un poliglucopéptido de fusión viral de clase I, es escindida por la proteasa similar a la furina del huésped en dos dominios funcionales, S1 y S213. La subunidad S1 contiene un dominio de unión al receptor de la célula huésped, mientras que la subunidad S2 se encarga de la fusión del virus a las membranas celulares15. La región del dominio de unión al receptor contiene un residuo de glutamina (Gln-394) que es reconocido por el residuo crítico de lisina (Lys-31) del receptor de la enzima convertidora de angiotensina 2 (ACE2) y que permite el ingreso del virus al interior celular16.
Mecanismos patogénicos de SARS-CoV-2
Mecanismo de infección y replicación viral
Una vez que el SARS-CoV-2 alcanza el organismo, inicia la replicación viral primaria en la mucosa epitelial del tracto respiratorio superior. A partir de allí, es capaz de invadir a las células epiteliales bronquiales y alveolares, macrófagos pulmonares, el endotelio vascular, la mucosa gastrointestinal y otros órganos17 a través de la interacción proteína S/ACE2. Así, a partir de los días 8-10 del inicio de la infección pueden aparecer los síntomas asociados al síndrome de distrés respiratorio agudo (SDRA) y la clínica extrapulmonar18-20.
La estructura de la región S1 de la proteína S del SARS-CoV-2 está compuesta por dos dominios independientes: el dominio N-Terminal (NTD) y el C-terminal (C-Domain). La invasión celular se produce mediante la interacción entre los residuos peptídicos A475-S19, N487-Q24, E484-K31 y Y453-H34 del C-Domain y el receptor para ACE2 mediante fuertes enlaces polares21,22. Tras formación del complejo proteína S/ACE2, la serina-proteasa transmembrana-2 (TMPRSS2) y la Catepsina L y B de las células endoteliales ceban la proteína S para luego ser escindida en el sitio de unión transmembrana por proteasas similar a la furina23 y permitir su entrada en el interior de la célula (Fig. 1).
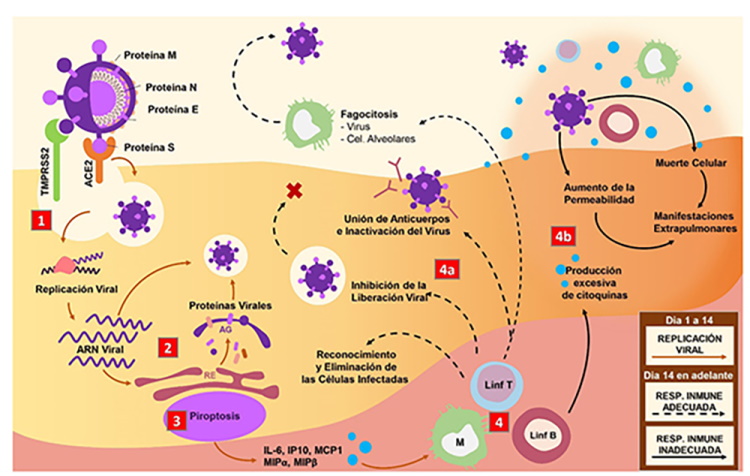
ACE2: enzima convertidora de angiotensina 2; TMPRSS2: proteasas de serina transmembrana 2; ARN: ácido ribonucleico; RdRp: ARN polimerasa dependiente de ARN; Mpro: proteasa principal viral; AG: aparato de Golgi; RE: retículo endoplasmático; IL-6: interleucina 6; IP-10: proteína 10 inducida por interferón gamma; MCP1: proteína quimiotáctica de monocitos 1; MIPα: proteína inflamatoria de macrófagos α; MIPβ: proteína inflamatoria de macrófagos β; M: monocitos/macrófagos; Linf: linfocitos; NAb: anticuerpos neutralizadores; SDRA: síndrome de distrés respiratorio agudo
Figura 1. Mecanismo patogénico del SARS-CoV-2. 1. El complejo proteína S/ACE2/TMPRSS2 activa una vía de señalización que facilita la entrada del virus. 2. El ARN viral es liberado en el citoplasma y se inicia la replicación viral mediada por RdRp. La proteasa viral Mpro da lugar a un gran número de proteínas víricas. 3. La replicación viral desencadena el proceso de piroptosis, liberación de citoquinas y quimiocinas proinflamatorias, lo cual permite la atracción de monocitos y linfocitos T y B. 4. La respuesta secundaria ocurre de manera adecuada (4a) con inactivación de virus mediada por NAb, reconocimiento y fagocitosis de las células infectadas, o de manera inadecuada (4b): se establece una tormenta de citoquinas, SDRA y aparición de manifestaciones extrapulmonares.
Una vez dentro de la célula huésped, el SARS-CoV-2 pierde su envoltura, su genoma se libera en el citoplasma y se inicia la replicación del ARN viral por acción de una ARN polimerasa dependiente de ARN (RdRp)24. Una vez obtenidas las copias positivas, la proteasa Mpro da lugar a un gran número de proteínas víricas25.
Implicaciones del complejo proteína S/ACE2
En condiciones normales, la ACE2 hidroliza la Angiotensina (Ang) II en Ang-(1-7) y la Ang I en Ang-(1-9). Además, a partir de la hidrólisis de la Ang A (producto de la descarboxilación de la Ang II), o la descarboxilación del residuo de aspartato de la Ang-(1-7), se genera Alamandina. Estos productos antagonizan las acciones nocivas de la Ang II, produciendo vasodilatación y un estado antitrombótico, antinflamatorio, antifibrótico y antiproliferativo26. Por ello, se propone que durante la infección por SARS-CoV-2 el complejo proteína S/ACE2 disminuye la actividad de la ACE2, promoviendo un estado proinflamatorio, protrombótico y profibrótico en el individuo27,28 (Fig. 2).
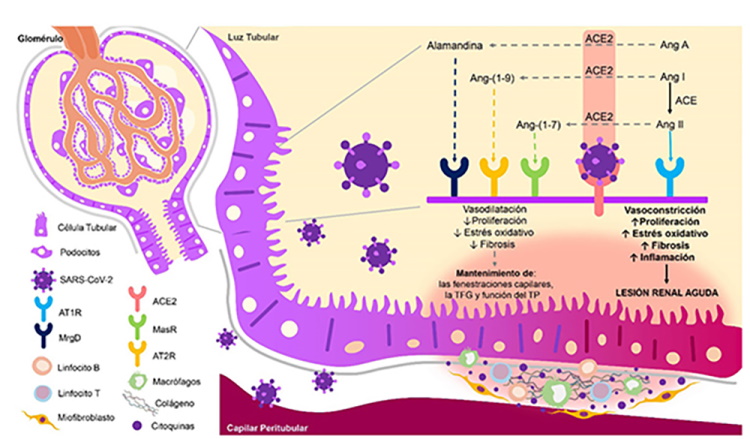
ACE2: enzima convertidora de angiotensina 2; SRA: sistema renina-angiotensina; Ang: angiotensina; TFG: tasa de filtrado glomerular; TP: túbulo proximal; AT1R y AT2R: receptor de angiotensina tipo 1 y 2; MasR: receptor Mas; MrgD: miembro D del receptor acoplado a proteína G asociado a Mas.
Figura 2. Daño Renal Directo por SARS-CoV-2: Rol de la ACE2. En el riñón, la ACE2 se expresa en los podocitos y en la zona apical del borde en cepillo de las células de los túbulos proximales. Al invadir dichas células, el SARS-CoV-2 promueve el daño renal al favorecer una disminución en la expresión de la ACE2 y un aumento en la actividad del SRA, generando vasoconstricción, secreción de citoquinas y factores de crecimiento, infiltración intersticial de células inflamatorias, acumulación de matriz extracelular y fibrosis.
Mecanismos de respuesta inflamatoria
Las manifestaciones pulmonares y extrapulmonares de la COVID-19 se deben tanto a un efecto directo del virus como a un efecto indirecto del síndrome de liberación de citoquinas o tormenta de citoquinas29. Durante la respuesta inflamatoria primaria, previa a la aparición de anticuerpos neutralizadores (NAb)30 se puede producir una muerte celular programada altamente inflamatoria conocida como piroptosis31. Esta viene mediada por la escisión del segmento N-terminal de la gasdermina D (GSDMD), un regulador de la proliferación celular32. Secundariamente, se inicia una importante liberación de citoquinas proinflamatorias, patrones moleculares asociados a patógenos (PAMPs) y patrones moleculares asociados a daño (DAMPs)28,30-32.
Las citoquinas proinflamatorias involucradas en este fenómeno son el factor necrosis tumoral alfa (TNF-α), la interleucina 1 beta (IL-1β) y la interleucina 6 (IL-6)33-35. Esta última, ha recibido mucha atención desde el inicio de la pandemia por su potencial terapéutico. La IL-6 está asociada al desarrollo del SDRA e hiperinflamación33, mediante la formación de un complejo entre el receptor de IL-6 unido a membrana (mIL-6R) y la glucoproteína 130 (gp130), que da inicio a la cascada de señalización JAKs/STAT3 y a la activación de las células del sistema inmune innato (neutrófilos, macrófagos y células NK) y adquirido (linfocitos B y T). A su vez, se puede unir a la forma soluble de su receptor (sIL-6R), formando un complejo con un dímero de gp130, que activa la secreción del factor de crecimiento vascular endotelial (VEGF), proteína quimiotáctica de monocitos 1 (MCP-1) e interleucina 8, además de reducir la expresión de la cadherinas epiteliales de las células endoteliales. Estas señales promueven la permeabilidad vascular, el edema intersticial, la pérdida del tono vascular y la disfunción pulmonar28,33-37.
La respuesta inflamatoria secundaria se produce a través de la activación de receptores de tipo Toll (TLR7 y TLR8) ubicados en las células epiteliales y macrófagos alveolares. Estos receptores son estimulados por PAMPs, que incluyen fragmentos de ARN viral; y DAMPs, que incluyen partículas de ATP, ADN, oligómeros de la proteína tipo speck asociada a apoptosis (ASC), proteínas de alta movilidad del grupo 1 (HMGB1) y factores de coagulación32,34,36. Las proteínas reclutadas en este proceso inducen cascadas de señalización que concluyen con la activación del factor regulador de interferon (IRF), el factor nuclear kappa beta (NF-κβ) y la proteína activadora 1 (AP-1), que transcriben inteferones (IF) antivirales tipo I y III, y quimioquinas -como la monoquina inducida por IF gamma (MIG), la proteína 10 inducida por IF gamma (IP-10), y MCP-1-. Este estímulo quimiotáctico atrae a células (tales como macrófagos, células dendríticas y linfocitos B) que presentan antígenos virales a linfocitos T, guiados por una respuesta polarizada T helper 1 (Th1), que amplifica la secreción de citoquinas proinflamatorias como IL-1β, IL-6, TNF-α y el antagonista del receptor de interleucina 1 (IL1RA), empeorando el estado hiperinflamatorio34,36.
Lesión renal aguda y COVID-19
La evidencia disponible muestra que la COVID-19 (Tabla 1) puede afectar los tres compartimientos renales (vascular, glomerular y tubular)6,20,38-43. Se ha encontrado que tanto la citotoxicidad del virus, a través de la interacción entre el SARS-CoV-2 y la ACE2, como el estado hiperinflamatorio están asociados al empeoramiento del pronóstico renal. La vasodilatación sistémica y la elevación de la presión arterial pulmonar a consecuencia de los mediadores proinflamatorios y el SDRA pueden producir un incremento de la presión torácica e intraabdominal, hipoxemia, hipercapnia y acidosis. Todo ello puede reducir el flujo sanguíneo y promover la perdida de la respuesta vasodilatadora renal, aumentando el consumo de oxígeno en el túbulo renal proximal y disminuir la diuresis18,44.
Papel de la ACE2 renal
La expresión de gen de la ACE2 es hasta 100 veces mayor en el riñón que en el pulmón, lo cual puede explicar el daño renal presente en los pacientes con COVID-1945. Específicamente, la expresión de esta enzima es más elevada en la zona apical del borde en cepillo de las células del túbulo proximal y, en menor medida, en los podocitos, donde también se ha reportado la expresión del gen de la TMPRSS2, pero no en las células endoteliales ni mesangiales del glomérulo45,46. Asimismo, estudios anatomopatológicos han permitido identificar la presencia de antígenos del SARS-CoV-2 en lesiones difusas de los túbulos proximales, con pérdida del ribete en cepillo, presencia de vacuolas citoplasmáticas, oclusión de la luz microvascular -principalmente por eritrocitos- y otros cambios vasculares y glomerulares, lo que sugiere que el SARS-CoV-2 es capaz de infectar estas células e inducir de forma directa una LRA46,47.
La invasión de las células renales y la interacción del SARS-CoV-2 con su receptor promueve un desbalance de la actividad ACE/ACE2 que puede ser causa de daño renal como consecuencia del predominio de los efectos deletéreos de la Ang II sobre la vasculatura renal, el descenso en la tasa de filtrado glomerular y la disfunción túbular proximal28,48,49. El daño celular promueve la secreción de más citoquinas proinflamatorias y factores de crecimiento, infiltración intersticial de células inflamatorias y acumulación de matriz extracelular, lo que causa atrofia tubular y fibrosis intersticial (Fig. 2)50.
Tabla 1. Evidencia epidemiológica de la lesión renal aguda en pacientes con COVID-19
| Autor / Población | Diseño | N | Resultados | ||
|---|---|---|---|---|---|
| Prevalencia (%) | TRS (%) | Mortalidad (%) | |||
| Rubin y col38 | Prospectivo | 71 | 80 | 18 | 21 |
| Burdeos, Francia | |||||
| Hirsch y col43 | Retrospectivo | 5.449 | 33,6 | 14,3 | 35 |
| Nueva York, EEUU | |||||
| Chan y col39 | Retrospectivo | 3.993 | 46 | 19 | 50 |
| Nueva York, EEUU | |||||
| Xiao y col40 | Retrospectivo | 287 | 25,8 | ND | 4,2 |
| Wuhan, China | |||||
| Cheng y col20 | Retrospectivo | 701 | 5,1 | ND | 33,7 |
| Wuhan, China | |||||
| Li y col41 | Retrospectivo | 193 | 28 | 4 | 17 |
| Wuhan, China | |||||
| Wang y col6 | Retrospectivo | 138 | 3,6 | 1,5 | 4,3 |
| Wuhan, China | |||||
| Xu y col42 | Retrospectivo | 355 | 15,8 | ND | 33,9 |
| Wuhan, China | |||||
TRS: tratamiento renal sustitutivo; ND: no disponible.
Aunque los resultados son controvertidos, la evidencia preclínica sugiere que la pérdida de la actividad de la ACE2 podría conferir protección contra la infección por coronavirus. No obstante, el bloqueo de la ACE2 pareciera aumentar su expresión y podría contribuir en la hiperactivación del sistema renina-angiotensina (SRA) y la exacerbación de la respuesta inflamatoria, empeorando así el curso de la enfermedad28,51-53. A pesar de esto, no hay evidencia sobre los efectos deletéreos del uso de fármacos que bloquean el SRA en los pacientes con COVID-19, por lo que su impacto debe ser estudiado con mayor profundidad54.
Tormenta de citoquinas y LRA
En un intento de comprender los patrones de respuesta inmune ante la infección por SARS-CoV-2, Mathew y col55 identificaron tres inmunotipos diferentes: 1) un grupo de individuos presentaban una robusta activación y proliferación de las células T CD4+, en ausencia relativa de células T helper foliculares (Tfh), junto a la marcada activación y agotamiento de las células T CD8+, y presencia de plasmoblastos (PB) T-bet+; 2) un segundo grupo con subpoblaciones de células T CD8+ efectoras más tradicionales, una respuesta menos robusta de las células T CD4+ y proliferación de PB y células B de memoria; y 3) un último inmunotipo caracterizado por ausencia de respuesta linfocítica, que sugiere un fallo de activación inmune, relacionando al inmunotipo 1 con una forma más severa de la enfermedad.
Asimismo, en pacientes hospitalizados con COVID-19, se ha observado un estímulo prolongado de las células T CD4+, CD8+ y PB a causa de un defecto en la regulación a la baja de su respuesta inmune. Esto podría resultar en el desarrollo de la hiperinflamación característica de la tormenta de citoquinas, capaz de generar rabdomiolisis y daño multiorgánico (Fig. 3)55. Es más, se ha correlacionado la magnitud de este proceso con la severidad de la enfermedad, donde los niveles de proteína C reactiva, TNF-α, IL-6, ferritina y creatina-fosfoquiinasa al momento de la hospitalización han demostrado ser significativamente más elevados en los pacientes que no han sobrevivido al curso de la enfermedad48,56.
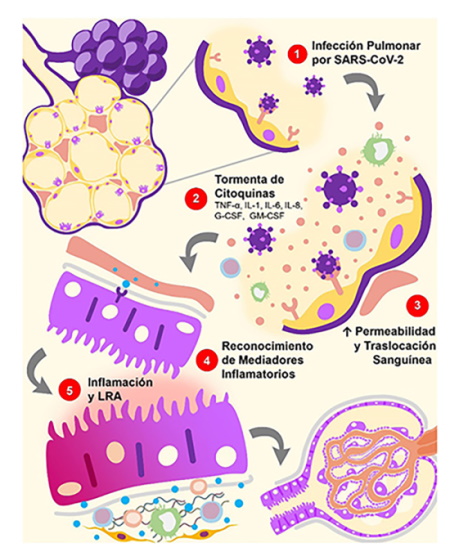
LRA: lesión renal aguda; IL: interleucina; TNF-α: factor de necrosis tubular alfa; PRR: receptor de reconocimiento de patrones; G-CSF: factor estimulante de colonias de granulocitos; GM-CSF: factor estimulante de colonias de granulocitos y macrófagos.
Figura 3. Daño Renal Indirecto por SARS-CoV-2: Inflamación Sistémica. 1. La infección por SARS-CoV-2 es capaz de generar una respuesta excesiva de las células T efectoras y producción aberrante de citoquinas proinflamatorias. 2. Este fenómeno es conocido como tormenta de citoquinas, proceso implicado en los procesos de coagulación, adhesión y permeabilidad vascular. 3. Una vez en la circulación sistémica, las citoquinas proinflamatorias pueden generar disfunción multiorgánica. 4. A nivel renal, los mediadores inflamatorios interactúan con los PRR presentes en las células inmunes y no inmunes. 5. Esto favorece un estado proinflamatorio y ejerce efectos deletéreos en las células de los túbulos renales.
Estos mediadores inflamatorios pueden contribuir al desarrollo de LRA al interactuar con las células inmunes y no inmunes, causando daño tubular y endotelial (endotelitis). Tal es el caso del TNF-α que, al actuar sobre su receptor (TNFR1), es capaz de generar: 1) apoptosis de las células tubulares; 2) pérdida de las fenestraciones glomerulares al disminuir la expresión del VEGF y de su receptor (sFle-1) en podocitos y células endoteliales; 3) degradación de la capa superficial endotelial, al aumentar la expresión de la heparenasa glomerular; 4) e infiltración de neutrófilos en el parénquima renal57 que, a su vez, puede ser promovida por la acción de la IL-6 y la IL-1β58.
Asimismo, los diferentes PAMPs y DAMPs ejercen efectos nocivos en los túbulos renales, mientras que las alteraciones vasculares generan focos de hipoperfusión e hipoxia que amplifican el proceso inflamatorio y causan mayor estrés oxidativo. Todo esto conlleva al desarrollo de LRA que puede agravar el daño presente en los pulmones y otros órganos, e incluso desencadenar la muerte48.
Activación del complemento y LRA
La activación inadecuada del sistema de complemento es capaz de generar un estado proinflamatorio crónico local y/o sistémico, muerte celular y daño multiorgánico en pacientes con COVID-1959. Según los datos disponibles, los residuos de aminoácidos (115-123) presentes en el dominio N-terminal de la proteína N del SARS-CoV-2, interactúan con la región C-terminal CCP1-CCP2-SP de la serina proteasa-2 asociada a la proteína de unión a manosa (MASP-2), dando inicio a una activación descontrolada de la vía del complemento activada por lectina, caracterizada por un aumento de la escisión de C4 y deposición del complemento en diferentes órganos59,60.
En el tejido renal, se ha observado la presencia de proteína N en las células tubulares, mas no en las glomerulares, y partículas citoplasmáticas similares a virus de aproximadamente 80-160 nm aportando evidencia de la capacidad de SARS-CoV-2 de producir un efecto citotóxico directo, además de a la respuesta inmune desencadenada a nivel local, al favorecer la infiltración tubulointersticial de macrófagos y una marcada acumulación del complejo de ataque a la membrana (también conocido como C5b-9) a nivel tubular, glomerular y vascular, lo que podría desencadenar necrosis tubular y LRA59,60.
Trombosis y LRA
Ante la infección por SARS-CoV-2 se genera un ambiente protrombótico, principalmente en pacientes gravemente enfermos, caracterizado por el desarrollo de eventos trombóticos venosos o arteriales. En este contexto, se ha descrito un estado de hipercoagulabilidad, lesión endotelial y estasis vascular como consecuencia de la interacción de la respuesta inmune innata con los diferentes elementos de la cascada de coagulación61,62.
La lesión endotelial puede estar mediada por citotoxicidad directa del SARS-CoV-2 sobre el endotelio vascular renal, o por lesión secundaria a la respuesta inflamatoria. Esta última se produce por activación de macrófagos y neutrófilos, formación de especies reactivas de oxígeno, disfunción endotelial, vasoconstricción e isquemia. Este ambiente protrombótico puede alterar el flujo de los vasos renales, así como promover la formación de trombos de fibrina a nivel glomerular, arteriolar y de los capilares peritubulares, contribuyendo al desarrollo de LRA45,48,63,64. A esto se suma una elevación en los niveles del factor de vonWillebrand, activación plaquetaria y del complemento, inhibición de la fibrinólisis y la producción de trampas extracelulares de neutrófilos (NET), que favorecerían la formación de microtrombos64,65.
Enfermedad renal crónica y COVID-19
Los pacientes con ERC previa pueden desarrollar un empeoramiento de la función renal o una lesión renal secundaria a la infección por SARS-CoV-2. Estos pacientes, ya sean dependientes o no de terapia renal sustitutiva (trasplante renal o diálisis) asocian una mayor mortalidad1,20,66.
A nivel tubular, se ha descrito una afectación proximal, o síndrome de Fanconi, que con frecuencia evoluciona hacia una LRA con un patrón de lesión tubulointersticial. Además, la LRA podría no resolverse, conduciendo a una insuficiencia renal crónica secundaria67,68.
En el compartimiento glomerular, la hiperinflamación y la endotelitis pueden condicionar una microangiopatía trombótica. Incluso se ha descrito glomerulopatía colapsante en portadores de polimorfismos de la apoliproteína-L1. En estos casos, el SARS-CoV-2 podría activar la vía del interferón-quimiocina CXCL9, causando una interrupción de la autofagia y la homeostasis mitocondrial, una linfohistiocitosis hemofagocítica y, finalmente, la muerte de las células glomerulares67-69.
Además, se ha señalado la presencia de hiponatremia asociada a secreción inadecuada de vasopresina, así como un síndrome cardiorrenal secundario a la hipoxemia y la hiperinflamación67,68. Pese a ello, un grupo de pacientes en diálisis presentó un curso más benigno70, probablemente debido a una respuesta inmune crónicamente disminuida (reducción en los niveles de linfocitos T colaboradores y citotóxicos, células NK y niveles bajos de citoquinas proinflamatorias), lo que podría favorecer el pronóstico70.
En conclusión, la evidencia disponible sostiene la hipótesis sobre el rol del SARS-CoV-2 en el desarrollo de nefropatía aguda o crónica; así como un aumento en la mortalidad. Esto podría deberse a la citoxicidad directa del virus o a la tormenta de citoquinas durante la respuesta inmune. En contraste, algunos casos de ERC, en diálisis y no en diálisis, presentan un curso más benigno, probablemente por una menor respuesta inflamatoria. Es por ello que son necesarios más estudios preclínicos y clínicos que permitan conocer el verdadero impacto de la COVID-19 y sus implicaciones a largo plazo en el pronóstico renal, así como dilucidar el rol de comorbilidades frecuentes en los individuos con ERC, como la anemia o la enfermedad óseo-mineral.

