










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRCOE
versión impresa ISSN 1138-123X
RCOE vol.9 no.4 jun./ago. 2004
| Eritema multiforme.
|
|

Erythema multiforme. Revision and update
Gavaldá-Esteve, Carmen*
Murillo-Cortés, Judith**
Poveda-Roda, Rafael**
*Profesora Asociada de Medicina Bucal. Universidad de Valencia.
** Médico Adjunto del Consorcio Hospital General Universitario de Valencia
Correspondencia
Carmen Gavaldá Esteve
C/ Caravaca, 18 pta. 8
46021- Valencia.
E-mail: cgavaldaesteve@yahoo.es
Resumen: El eritema multiforme es una enfermedad de la piel y las mucosas que se manifiesta con lesiones eritematosas y de tipo vesículo-ampollar. Las lesiones vesiculo-ampollares y erosivas a nivel de la cavidad oral y la piel pueden ser causadas por un amplio grupo de patologías. La etiología de las mismas también puede ser muy variable, desde una causa traumática o química por contacto, hasta una causa autoinmune. Dado que en ocasiones es difícil hacer un diagnóstico diferencial para discernir la etiología de las lesiones, es importante conocer los detalles clínicos y los aspectos epidemiológicos e histopatológicos de cada una de ellas.
En este artículo se hace una revisión de los aspectos epidemiológicos, etiopatogénicos, clínicos, histopatológicos, de tratamiento y pronóstico del eritema multiforme.
Palabras clave: Eritema multiforme, Mucosa oral.
Abstract: Erythema multiforme is a disorder of the skin and mucosal membranes, manifesting as erythematous and vesicle-blister lesions. In the oral cavity, the vesicle-blister and erosive lesions can be caused by a considerable variety of disorders. The underlying etiology is highly variable, and ranges from traumatisms or contact chemical action to autoimmune mechanisms. Since a differential diagnosis is sometimes difficult to establish, it is important to know the clinical, epidemiological and histological particularities of each of the possible causes.
A review is made of the epidemiological, etiopathogenic, clinical, histological, management and prognostic characteristics of erythema multiforme.
Key words: Erythema multiforme, Oral mucosa.
BIBLID [1138-123X (2004)9:4; julio-agosto 361-476]
Gavaldá-Esteve C, Murillo-Cortés J, Poveda-Roda R. Eritema multiforme. Revisión y puesta el día. RCOE 2004;9(3):415-423.
Introducción
El eritema multiforme (EM) o eritema polimorfo es una enfermedad aguda de la piel y/o de las mucosas de naturaleza inmunológica, que puede comportarse como crónica recurrente, y que se caracteriza por presentar lesiones cutáneas eritemato-bullosas de varios tipos y/o lesiones mucosas pluriorificiales de tipo vesículo-ampollar1-6.
En cuanto a la clínica, clásicamente se distingue entre una forma menor o recurrente y las formas mayores que incluyen el síndrome de Stevens-Johnson (SSJ) y el síndrome de Lyell o necrólisis epidérmica tóxica (NET)1,5,7-9, aunque hay autores que las consideran entidades diferentes10,11**-13*.
Epimediología. Etiopatogenia
La aparición del EM puede darse a cualquier edad con un pico de incidencia en pacientes jóvenes, sobre todo entre la segunda y tercera décadas de la vida1,2,14. También puede aparecer en niños aunque es raro, sobre todo, en la primera infancia14,15.
La incidencia del EM mayor es según Nanda et al15 de 0,8-6,0 por millón y año.
Predomina ligeramente en el sexo masculino1,8,12 con una proporción que oscila entre 3:2 a 2:115 y no parece existir predilección racial16.
Su etiología es desconocida aunque en muchos casos aparece vinculado a factores predisponentes como infecciones por virus (tabla 1). El más frecuente es el herpes simple (VHS), pero también por hepatitis o mononucleosis infecciosa5. Se ha demostrado por métodos de reacciones de polimerasa en cadena (PCR) la presencia de DNA del VHS en las lesiones de la piel y también se ha hallado el DNA viral en las células epiteliales por métodos de hibridación in situ17. Asimismo, se han descrito como factores desencadenantes la infección por Mycoplasma pneumoniae y la ingesta de determinados fármacos1,2,4,7,9,12,18*-21.

El herpes es el principal factor de riesgo del EM menor (forma recurrente) y se estima que entre el 15 y el 63% de los casos de EM son secundarios al VHS y que la mayoría de casos considerados hasta ahora como idiopáticos estarían relacionados con infecciones subclínicas por VHS5. Los fármacos se asocian sobre todo con las formas mayores (síndrome de Stevens-Johnson y síndrome de Lyell)12,18*, aunque también el EM menor puede estar desencadenado por fármacos8. No hay evidencia objetiva de que la dosis del medicamento influya en el grado de intensidad del EM8,22. El número de fármacos que pueden provocar el cuadro es muy amplio y continuamente se describen nuevos casos de EM relacionados con los mismos, por lo que es un listado siempre abierto a nuevas aportaciones (tabla 2).

Además de los descritos en la tabla 2 se han publicado entre otros:
- Casos de EM y SSJ con el uso de Bupropion (Zyntabac®), utilizado como antidepresivo y para dejar de fumar8.
- Rodríguez Vázquez y col4 reportan el caso de un EM menor en una mujer de 39 años inducido por tetracepam (Myolastan®) y dicen que también puede producir, al igual que otras benzodiacepinas, un SSJ.
- Nanda et al15 informan de un caso de EM bulloso en una niña de nueve días tras la ingesta dos días antes de un tratamiento homeopático para un resfriado común.
- Han sido publicados algunos casos en los que la asociación de un fármaco anticonvulsivante (fenitoína) y radioterapia craneal ha desencadenado un brote de EM22.
- También con terapias antirretrovirales para el tratamiento de pacientes VIH+, como Zidovudina (AZT), Didanosina (DDI), Zalcitabina (DDC) o Abacavir (ABC), se han publicado casos de eritema multiforme con elevado porcentaje de afectación oral para algunos de ellos20.
- Se han descrito casos de NET con ofloxacino, ciprofloxacino y trovafloxacino (antibióticos quinolónicos)19. Para este autor los fármacos implicados en la NET, en orden decreciente de riesgo relativo, son: Sulfonamidas, clormezanona, antiinflamatorios no esteroideos, antifúngicos imidazólicos, cefalosporinas, anticonvulsivantes y alopurinol. Otras posibles causas implicadas en la NET y EM son: inmunizaciones, infecciones víricas, enfermedad injerto contra huésped, enfermedades malignas hematológicas, lupus eritematoso sitémico, poliarteritis nudosa, tumores cerebrales, e infecciones del tracto respiratorio superior5,19.
- También se han publicado casos en los que la progesterona endógena actúa como antígeno, provocando brotes de EM con cada ciclo menstrual23 y casos de reacciones a píldoras anticonceptivas2.
- Por otra parte, la fotodistribución de las lesiones, o aumento en la densidad y confluencia de las mismas, en zonas de la piel expuestas al sol es un fenómeno descrito en relación al EM. También se han publicado casos en los que una infección por virus del herpes simple o la ingestión de fármacos previa a la exposición solar, han desencadenado un brote de EM, incluso se han descrito dos casos en los que la radiación solar por sí sola ha provocado un cuadro de EM, afectando también zonas de la piel no expuestas24.
- Como hemos comentado antes pueden aparecer casos coincidentes de EM y lupus eritematoso sistémico. Si además el paciente presenta un patrón moteado de anticuerpos antinucleares (ANA), factor reumatoide positivo y anticuerpos precipitantes al extracto salino de tejido humano (anti-Sj-T), nos encontramos ante un síndrome de Rowell. Es un síndrome raro descrito por Rowell et al en 196325.
Patogénicamente, aunque se acepta que las lesiones del EM son debidas a una reacción de hipersensibilidad a un antígeno (principalmente agentes microbianos o fármacos), para algunos autores16,17,26,27 se trata de una reacción tipo III mediada por inmunocomplejos que provocarían una vasculitis y secundariamente una necrosis isquémica del epitelio, mientras que para otros5,8,21,26 se trata de una reacción tipo IV mediada por células (linfocitos T), que producen la necrosis en mayor o menor grado de las células epidérmicas. La epidermis sufre una reacción citotóxica en la que el fármaco que ocasiona la reacción o alguno de sus metabolitos se une a las proteínas de membrana de los queratinocitos, convirtiéndoles en blanco del ataque celular13*. La apoptosis parece ser el mecanismo por el que las células citotóxicas inducen la muerte celular epidérmica13*. También se ha descrito una mayor susceptibilidad para el EM en las personas que presentan el HLA-DQB11,26. En ocasiones se han implicado factores metabólicos13*.
Clínica
El eritema multiforme fue descrito por primera vez por von Hebra en 1866 como un cuadro clínico donde aparecían lesiones cutáneas con cambios concéntricos de color (en diana o escarapela), simétricamente distribuidas. En 1922 Stevens y Johnson publicaron dos casos de niños con exantema generalizado con lesiones cutáneas distintas a las descritas por von Hebra y afectación de mucosas oral y oftálmica así como fiebre. En 1950 Thomas sugirió que el EM y el SSJ eran variantes del mismo proceso patológico, y propuso llamar EM menor a la forma cutánea menos agresiva descrita por Hebra y EM mayor a las formas cutáneo-mucosas más agresivas decritas por Stevens y Johnson. En 1956, Lyell publicó una serie de pacientes con una reacción cutáneomucosa grave que incluía un extenso eritema que evolucionaba rápidamente a necrosis, grandes ampollas con despegamiento dermo-epidérmico y que tenía un grave pronóstico. Es un cuadro que se conoce como necrólisis epidérmica tóxica11**.
Actualmente existe controversia en cuanto a la clasificación clínica de la enfermedad. Gran parte de autores1,5-9,14 siguen clasificando al EM, al SSJ y NET como expresiones distintas de una misma entidad clínico-patológica, diferenciando, como ya dijimos, entre formas menores y mayores, que varían en cuanto a extensión, gravedad, y posible factor desencadenante (virus vs fármacos). Otros autores sin embargo consideran que el EM es una entidad diferente del SSJ y NET. Estas diferencias incluyen aspectos clínicos, etiológicos e histopatológicos10,11**,13*.
En cuanto a la afectación de la mucosa oral en el EM también existen discrepancias. Así, aunque la entidad descrita por von Hebra no incluía afectación oral, la mayoría de los autores aceptan que pueden existir ocasionalmente lesiones eritematosas o erosivas a nivel oral formando parte del EM8,11**, y varios autores describen lesiones orales típicas de EM como única o como manifestación principal del cuadro clínico de EM11**. Otros autores sin embargo opinan que no está justificado el diagnóstico de EM si no hay lesiones en piel, siendo las lesiones cutáneas típicas condición sine qua non para el diagnóstico de EM11**.
Ayangco y Rogers en el 200311**, realizaron una clasificación clínica del EM teniendo en cuenta las clasificaciones anteriores y las manifestaciones a nivel oral. Distinguen: EM menor, EM mayor, SSJ, NET y EM oral, con características clínicas, de evolución y de pronóstico diferentes pero que comparten dos características comunes: lesiones cutáneas en diana típicas o atípicas (los cuatro primeros) y necrosis epitelial de extensión variable. Aunque no es una clasificación aceptada por todos los autores, nos servirá de guía para describir a continuación las características clínicas de las lesiones cutáneas y mucosas.
Eritema multiforme menor
Lesiones agudas recurrentes o no, autolimitadas. Curan en 2-4 semanas sin secuelas.
Piel: lesiones en diana o escarapela típicas de menos de 3 mm de diámetro, forma redondeada y regular, borde bien definido y con al menos tres zonas concéntricas diferentes, o lesiones en diana atípicas, con tan sólo dos zonas concéntricas o bordes mal definidos11**. Puede haber vesículas en la zona central14.
Localizadas generalmente de forma simétrica en superficies extensoras de las extremidades o en la cara, y menos frecuentemente en las palmas y plantas1,2,7,12,14, afectando menos del 10% de la superficie corporal y con signo de Nikolsky negativo11**.
Mucosas: afectación mínima o inexistente de las mucosas en forma de máculas eritematosas o erosiones superficiales de la mucosa oral y los labios11**.
Eritema multiforme mayor
Lesiones agudas recurrentes o no, autolimitadas. Al igual que en el EM menor, las lesiones curan en 2-3 semanas (pueden persistir hasta seis según Lineberry et al8 y Nanda et al15) sin dejar secuelas pero pueden producirse nuevos brotes al cabo de un periodo variable por lo que se considera una enfermedad crónica manifestada por múltiples brotes agudos1-3. Suelen aparecer tras días o semanas tras la exposición con el antígeno (brote de herpes recidivante, ingesta de fármacos)2,8.
Piel: lesiones iguales a las del EM menor pero más extensas, con signo de Nikolsky negativo..
Mucosas: afectación de una o más mucosas (generalmente la de la cavidad oral), que pueden ser la manifestación más importante del cuadro clínico. Del 40% al 60% de los pacientes tienen lesiones orales que se localizan típicamente en la parte anterior de la cavidad oral y lengua, en mucosa no queratinizada, siendo infrecuente la afectación gingival6,11** aunque puede ocurrir aproximadamente en el 16% de pacientes con lesiones orales16. Clínicamente las lesiones son variables, encontrando zonas eritematosas, máculas hiperémicas, pápulas o vesiculoampollas y erosiones superficiales cubiertas por una pseudomembrana de fibrina (figs. 1-4).




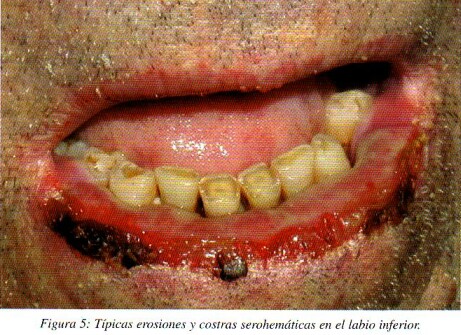
Pueden aparecer lesiones en diana en los labios, que además presentan de forma típica erosiones y costras serohemáticas (fig. 5). Las lesiones mucosas pueden preceder, aparecer simultáneamente, o posteriormente a las lesiones en piel y curan sin cicatriz. Marinho et al3. presentan un caso excepcional en el que tras un brote de EM mayor, un niño de ocho años presentó cicatrices en las comisuras labiales que le impedían la correcta apertura bucal. Las lesiones cutáneas pueden dejar tras su curación una zona hiperpigmentada de larga duración8. En ocasiones aparecen linfadenopatías cervicales1,6,11**,14,16,18*.
Síndrome Stevens-Johnson
Lesiones agudas no autolimitadas. Es una enfermedad grave con mal estado general que incluye manifiestaciones viscerales además de las de la piel y las mucosas1,7,12,14.
Piel: pequeñas vesículas diseminadas por la piel que evolucionan hacia máculas purpúricas o lesiones en diana atípicas, localizadas sobre todo en el torso más que en las extremidades, con despegamiento epidérmico en algunas zonas que no supera el 10% de la superficie corporal. Signo de Nikolsky positivo.
Mucosas: afectación de una o más mucosas (oral, ocular, nasal, genital) con secuelas cicatriciales. Se afectan típicamente la mucosa bucal y el paladar con ampollas y úlceras profundas cubiertas por pseudomembranas, así como el bermellón de los labios con abundantes erosiones y costras serohemáticas. Son lesiones muy dolorosas. En casos muy severos las lesiones pueden afectar a: encías, lengua, faringe, mucosa nasal, laringe, esófago y árbol respiratorio1,7,11**,12.
En un 40% de los casos se afectan también las mucosas anogenital, palpebral y conjuntiva. Las lesiones curan con cicatrices, sobre todo a nivel ocular (produciendo incluso ceguera) y en garganta, esófago, bronquios y mucosa anogenital11**,14.
Necrólisis epidérmica tóxica
Lesiones agudas no autolimitadas, sino progresivas. Afectación de la piel, las mucosas y órganos internos con un curso fulminante. Es una enfermedad extremadamente grave1,7,12. La NET típica desencadenada por un fármaco ocurre a las tres semanas tras iniciar la toma del mismo19, aunque puede ocurrir pocos días después si el paciente ya había estado en contacto con el fármaco26.
Piel: afectación similar al SSJ pero más extensa, con formación de ampollas y despegamiento epidérmico del 30% hasta el 100% de la superficie corporal. Signo de Nikolsky positivo11**,19.
Mucosas: afectación similar a la descrita en el SSJ pero más severa que también cura con cicatrices1,7,11**,12.
Eritema multiforme mayor oral
Lesiones agudas recurrentes o no, o crónicas durante periodos de tiempo prolongados (semanas o meses).
Piel: lesiones típicas de EM menor pueden hallarse en el 25% de los pacientes.
Mucosas: Eritema, ampollas intraorales y erosiones con o sin pseudomembrana, o placas hiperqueratósicas no específicas entremezcladas con áreas eritematosas. Localizaciones variables, aunque se afectan con frecuencia la encía, y las mucosas labial y bucal y en menos ocasiones el bermellón de los labios11**.
Considerando todas las formas de EM como parte de una misma entidad, podemos decir que alrededor del 50% de los casos el paciente tiene unos síntomas prodrómicos inespecíficos parecidos a los de un resfriado común, astenia, fiebre, cefalea, artralgias1 que duran aproximadamente una semana o dos tras la exposición a un fármaco u otros estímulos antigénicos8 y que dan paso de forma brusca a la aparición de lesiones en la piel y/o la mucosa oral. Para Fernández-García y col5 y Lineberry et al8 los síntomas prodrómicos son raros en las formas menores y suelen darse en el SSJ y NET.
La incidencia de afectación oral varía considerablemente según distintos autores. Para algunos sólo el 25% de pacientes con afectación en piel tendrían lesiones orales, otros sin embargo reportan un 65%18, Farthing et al18* en un estudio sobre 82 pacientes encontraron un 70% de afectación oral, por tanto de acuerdo con Ayangco y Rogers11**, y Bowers et al14 reportan afectación de la mucosa oral entre el 40%-60% de los casos o entre el 35% y 65% respectivamente. De forma exclusiva puede darse hasta en un 43% de los casos1.
Diagnóstico
El diagnóstico se realiza sobre todo por la clínica2 combinado con la histología, cuyos hallazgos no son patognomónicos17. En el estudio histológico encontraremos edema intra e intercelular, queratinocitos necróticos con licuefacción focal en las capas inferiores de la epidermis, infiltrado inflamatorio mixto con predominio de linfocitos T a lo largo de la unión dermoepidérmica, alteración vacuolar de la capa basal y fisuras en la unión dermoepidérmica con formación de ampollas subepiteliales y en ocasiones intraepiteliales así como infiltrado linfohistiocitario perivascular sin vasculitis. La inmunofluorescencia tanto directa como indirecta es negativa y sirve sobre todo para descartar otros procesos1,5-7,9,11**,14,16,17,27.
Los hallazgos de laboratorio en el EM menor son normales. En el EM mayor puede haber una tasa de sedimentación globular aumentada, leucocitosis moderada y ligero aumento de transaminasas. En el SSJ y NET, hay fiebre y los hallazgos de laboratorio dependerán del grado de afectación de los órganos internos11**.
Debe hacerse un diagnóstico diferencial con otras entidades como la primoinfección herpética, sobre todo si hay ausencia de lesiones en la piel, estomatitis aftosa recidivante, pénfigo, pénfigo paraneoplásico y penfigoide11**,18*.
Tratamiento
Depende de la severidad de la enfermedad. Ante manifestaciones leves se administra tratamiento sintomático de las lesiones, como analgésicos tópicos o sistémicos, compresas con suero, enjuagues bucales con anestésicos, dieta blanda, evitar comidas picantes y aumentar la ingesta de líquidos1,5,11**,15,21. También pueden administrarse corticoides tópicos y antibioterapia para prevenir infecciones3,11**. En pacientes adultos con historia de infección por VHS, una terapia antivírica puede producir beneficios, sobre todo previniendo recurrencias cuando se aplica de forma profiláctica2. El tratamiento del episodio agudo con aciclovir sólo está indicado cuando se instaura muy precozmente21. Las dosis varían algo según distintos autores, Fernández-García y cols5 proponen 200-800 mg de aciclovir/día para ir disminuyendo progresivamente hasta encontrar la dosis menor capaz de prevenir las recidivas, Hernanz-Hermosa y cols21 dan 200-400 mg/día durante periodos prolongados de cinco o seis meses, Bowers et al14 y Farthing et al18* pautan 400 mg dos veces al día de aciclovir, sin especificar la duración del tratamiento.
Existe controversia respecto al uso de corticoides sistémicos para el tratamiento del EM, sobre todo en cuadros clínicos moderados2,3,9 y los EM recurrentes debidos al herpes simple. La instauración del tratamiento iría encaminada a mejorar la sintomatología y acortar el proceso1, pero parece ser que la tendencia actual es no utilizarlos5,11** pues no está demostrado que acorte la duración de la enfermedad9 y pueden estar asociados a un aumento en la frecuencia de los brotes de EM y su cronicidad18*. Las dosis que proponen los distintos autores también varían. Bagán1 recomienda dosis de 30-50 mg al día de prednisona o metilprednisolona durante varios días disminuyendo la dosis paulatinamente. Lineberry et al8 y León-Ruiz et al9 pautan 1 mg/Kg/día durante una semana y luego disminuyen progresivamente. Rodríguez-Vázquez y cols4 tratan el brote con la retirada del fármaco y prednisona 30 mg/día durante dos semanas. Barret et al16 pautan 30 mg de prednisolona diaria durante 10 días y la van reduciendo hasta llegar a dosis de 10 mg a días alternos. El tratamiento con corticoides está contraindicado en pacientes con una función inmune comprometida o procesos infecciosos premórbidos en los cuales la inducción de una inmunosupresión por parte de los corticoides podría ser peligrosa. La tuberculosis podría ser un ejemplo de infección problemática8.
Otros autores obtienen buenos resultados con 150 mg al día de levamisol durante tres días consecutivos solos o asociados con corticoides28,29. Otros fármacos reportados como efectivos en EM recurrentes refractarios a otros tipos de tratamiento son la azatioprina (100-150mg/día), la talidomida14,18*, la dapsona, el metotrexate y el micofenolato11**. Un pequeño porcentaje de pacientes han obtenido buena respuesta con fármacos antimaláricos como la hidroxicloroquina18*,24.
Ante un SSJ o NET el paciente precisa hospitalización en una unidad de quemados o de cuidados intensivos con control riguroso del equilibrio hidroelectrolítico y tratamiento sistémico con corticoides1,2,19 aunque este último también provoca controversia entre distintos autores13*.
En casos desencadenados por fármacos debe retirarse inmediatamente el fármaco sospechoso (sospecharemos si es un fármaco que se instauró entre 1 y 3 semanas antes y si es de los que ocasionan SSJ-NET con más frecuencia que otros13*). Ante requerimientos de fármacos esenciales se sustituirán por otros de similares efectos farmacológicos pero de diferente estructura química26.
Se realizará un tratamiento sintomático monitorizando la función respiratoria, hemodinámica y de nivel de conciencia. Es necesario controlar diariamente la superficie corporal con despegamiento epidérmico para valorar la necesidad de fluidos que se repondrán por vía intravenosa. Se extremarán las condiciones de antisepsia empleando antibióticos sistémicos sólo cuando sea necesario. Se puede hacer un desbridamiento de la piel necrótica y se extremarán los cuidados oculares para evitar secuelas y se utilizarán sprays de clorhexidina en la cavidad oral.
Además de los corticoides sistémicos se han utilizado inmunosupresores, plasmaféresis, agentes anticitocinas e infusión intravenosa de inmunoglobulinas aunque ninguno ha demostrado ser eficaz y existen dudas sobre la seguridad de muchos de ellos13*.
Pronóstico
En las formas menores de EM las lesiones resuelven espontáneamente en unas tres semanas sin secuelas. La instauración del tratamiento puede mejorar la sintomatología y acortar el proceso. En las formas mayores (SSJ y NET) los pacientes pueden fallecer o acabar con graves secuelas1. La tasa mortalidad para el SSJ se sitúa en torno al 10%11**, y para la NET se sitúa entre el 25-75% según distintos autores11,13,19,26. Empeoran el pronóstico la existencia de una gran extensión de zonas denudadas, la edad avanzada, la insuficiencia renal concomitante y la afectación pulmonar26.
Bibliografía recomendada
Para profundizar en la lectura de este tema, el/los autor/es considera/an interesantes los artículos que aparecen señalados del siguiente modo: *de interés **de especial interés.
1. Bagán-Sebastián JV. Enfermedades ampollares de la cavidad oral (III): Eritema multiforme, dermatosis IgA lineal, dermatitis herpetiforme y epidermólisis ampollar. En: Bagán-Sebastián JV, Ceballos-Salobreña A, Bermejo-Fenoll A, Aguirre-Urizar JM, Peñarrocha-Diago M, editores. Medicina Oral. Barcelona: Masson, 1995:234-40. [ Links ]
2. Ayangco L, Sheridan PJ, Rogers RS. Erythema multiforme secondary to herpes simplex infection: a case report. J Periodontol 2001;72:953-7. [ Links ]
3. Marinho LHM, Haj M, Pereira LFM. Lip adhesion. An unusual complication of erythema multiforme. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;88:167-9. [ Links ]
4. Rodríguez Vázquez M, Ortiz de Frutos J, del Río Reyes R, Iglesias Díez L. Eritema exudativo multiforme inducido por tetrazepam. Med Clin 2000;115:359. [ Links ]
5. Fernández García JR, Alcaraz Vera M, Ruiz Jiménez MA, Rodríguez Murillo JM, Hens Pérez A. Eritema multiforme. Rev Esp Pediatr 2000;56:202-5. [ Links ]
6. Holmstrup P. Non-Plaque-Induced Gingival Lesions. Ann Periodontol 1999;4:20-9. [ Links ]
7. Hernández G, Arriba de la Fuente L, Lucas M. Enfermedades ampollosas de la mucosa bucal. Características diferenciales clínicas, histológicas e inmunológicas. Medicina Oral 1999;4:528-51. [ Links ]
8. Lineberry MTW, Peters CGE, Bostwick JM. Bupropion-Induced Erythema Multiforme. Mayo Clin Proc 2001;76:664-6. [ Links ]
9. León Ruiz L, Morales Larios E, Díaz Ricomá N, Jiménez Alonso J. Úlceras orales y rash eritematopapuloso en varón de 17 años. Rev Clin Esp 2003;203:491-2. [ Links ]
10. Paquet P, Pierard GE. Erythema multiforme and toxic epidermal necrolysis: a comparative study. Am J Dermatopathol 1997;19:127-32. [ Links ]
11**. Ayangco L, Rogers RS. Oral manifestations of erythema multiforme. Dermatol Clin 2003;21:195-205. [ Links ]
Hace una revisión actualizada y amplia de los distintos aspectos clínicos, histológicos y de tratamiento del EM, centrándose sobre todo en las lesiones a nivel oral.
12. Auquier-Dunant A, Mockenhaupt M, Naldi L, Correia O, Schröder W, Roujeau JC. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis. Results of an international prospective study. Arch Dermatol 2002;138:1019-24. [ Links ]
13*. García Doval I, Roujeau JC, Cruces Prado MJ. Necrólisis epidérmica tóxica y síndrome de Stevens-Johnson: clasificación y actualidad terapéutica. Actas Dermosifiliogr 2000;91:541-51. [ Links ]
El artículo hace una revisión muy completa del SSJ y NET, en todos sus aspectos y de forma clara y sencilla.
14. Bowers KE. Oral Blistering Diseases. Clinics in Dermatology 2000;18:513-23. [ Links ]
15. Nanda S, Pandhi D, Reddy BSN. Erythema multiforme in a 9-day-old neonate. Pediatric Dermatology 2003;20:454-5. [ Links ]
16. Barrett AW, Scully CM, Eveson JW. Erythema multiforme involving gingiva. J Periodontol 1993;64:910-3. [ Links ]
17. Eversole LR. Immunopathology of oral mucosal ulcerative, desquamative, and bullous diseases. Selective review of the literature. Oral Surg Oral Med Oral Pathol 1994;77:555-71. [ Links ]
18*. Farthing PM, Maragou P, Coates M, Tatnall F, Leigh IM, Williams DM. Characteristics of the oral lesions in patients with cutaneous recurrent erythema multiforme. J Oral Pathol Med 1995;24:9-13
Destaca la descripción de lesiones de EM a nivel oral. [ Links ]
19. Melde SL. Ofloxacin: A Probable Cause of Toxic Epidermal Necrolysis. Ann Pharmacother 2001;35:1388-90. [ Links ]
20. Scully C, Diz Dios P. Orofacial effects of antiretroviral therapies. Oral Diseases 2001:7:205-10. [ Links ]
21. Hernanz JM, González-Beato M, Pico M, Pérez S, Marengo S. Eritema exudativo multiforme "minor". Acta Pediátrica Española 2000;58:89-90. [ Links ]
22. Ahmed I, Reichenberg J, Lucas A, Shehan JM. Erythema multiforme associated with phenytoin and cranial radiation therapy: A report of three patients and review of the literature. Int J Dermatol 2004;43:67-73. [ Links ]
23. Moghadam BKH, Hersini S, Barker BF. Autoinmune progesterone dermatitis and stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998;85:537-41. [ Links ]
24. Calzavara-Pinton PG, Venturini M, Capezzera R, Zane C, Facchetti F. Photosensitive erythema multiforme and erythema multiformelike polymorphous light eruption. Photodermal Photoimmunol Photomed 2003;19:157-9. [ Links ]
25. Pandhi D, Singal A, Agarwal P. Rowells syndrome and associated antiphospholipid syndrome. Clinical and Experimental Dermatology 2004;29:22-4. [ Links ]
26. Fernández Redondo V, Rosón López E, Gómez Centeno P. Toxicodermias: etiopatogenia, clínica y tratamiento. Medicine 1999;135:6367-73. [ Links ]
27. Chrysomali E, Lozada-Nur F, Dekker NP, Papanicolaou SI, Regezi JA. ¡OJO FALTA TÍTULO! Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:272-80. [ Links ]
28. Lu SY, Chen WJ, Eng HL. Response to levamisole and low-dose prednisolone in 41 patients with chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998;86:438-45. [ Links ]
29. Lozada-Nur F, Cram D, Gorsky M. Clinical response to levamisole in thirty nine patients with erythema multiforme. An open prospective study. Oral Surg 1992;74:294-8. [ Links ]

