










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkInternational Microbiology
versión impresa ISSN 1139-6709
INT. MICROBIOL. vol.7 no.3 sep. 2004
| RESEARCH ARTICLE | |||
|
| |||
| A new set of DNA macrochips for the yeast accharomyces cerevisiae: features and uses
Summary. The yeast Saccharomyces cerevisiae has been widely used for the implementation of DNA chip technologies. For this reason and due to the extensive use of this organism for basic and applied studies, yeast DNA chips are being used by many laboratories for expression or genomic analyses. While membrane arrays (macroarrays) offer several advantages, for many laboratories they are not affordable. Here we report that a cluster of four Spanish molecular-biology yeast laboratories, with relatively small budgets, have developed a complete set of probes for the genome of S. cerevisiae. These have been used to produce a new type of macroarray on a nylon surface. The macroarrays have been evaluated and protocols for their use have been optimized. [Int Microbiol 2004; 7(3):199-206] Key words: Saccharomyces cerevisiae · DNA chip · macroarray | ||
| |||
| Nuevos macrochips para el genoma completo de la levadura Saccharomyces cerevisiae: características y usos Resumen. La levadura Saccharomyces cerevisiae ha sido muy utilizada para el desarrollo de las tecnologías de chips de DNA. Por ese motivo, y porque es un organismo muy utilizado en investigación básica y aplicada, hay muchos laboratorios que usan chips de DNA para estudios genómicos o de expresión. Aunque el uso de macrochips en membrana presenta varias ventajas, su precio los pone fuera del alcance de muchos laboratorios. Aquí mostramos que un grupo de cuatro laboratorios españoles de biología molecular de levaduras ha desarrollado, con presupuestos relativamente bajos, un lote completo de sondas del genoma de S. cerevisiae, que se han usado para fabricar un nuevo tipo de macrochips sobre superficie de nailon. Se han evaluado estos macrochips y se han optimizado los protocolos para su uso. [Int Microbiol 2004; 7(3):199-206] Palabras clave: Saccharomyces cerevisiae · chip de DNA · macroseries (macroarray) | Novos macrochips para o genoma completo da levedura Saccharomyces cerevisiae: características e usos Resumo. A levedura Saccharomyces cerevisiae tem sido muito utilizada para o desenvolvimento das tecnologias de chips de DNA. Por esse motivo, e por se tratar de um organismo largamente empregado em pesquisa básica e aplicada, existem muitos laboratórios que usam chips de DNA para os estudos genômicos ou de expressão. Embora o uso de macrochips em membrana apresente várias vantagens, seu preço os coloca fora do alcance de muitos laboratórios. Aqui mostramos que um grupo de quatro laboratórios de biologia molecular espanhóis, desenvolveram com insumos relativamente baixos, um lote completo de sondas para o genoma de S. cerevisiae, que foram usadas para fabricar um novo tipo de macrochips sobre a superficie de nailon. Esses macrochips foram avaliados e otimizados os protocolos para seu uso. [Int Microbiol 2004; 7(3):199-206] Palavras chave: Saccharomyces cerevisiae · chip de DNA · macroseries (macroarray) |
Introduction
The yeast Saccharomyces cerevisiae has been used as a workhorse for the implementation of DNA chip technologies [8,22,26], and several different kinds of DNA chips were first assayed in yeast. For this reason, there are DNA chips with short (20- to 25-mers) [15] or long (60- to 70-mers) [16,25] oligonucleotide probes, and PCR-amplified double-stranded probes [7,8,11,20,25]. Apart from the popular glass-slide microarrays, macroarrays on positively charged nylon membranes have been developed [7,11,12].
Each type of DNA chip has its own advantages. PCR probes are more time-consuming but less expensive than oligonucleotides. Moreover, PCR probes have some additional advantages: they usually cover most of the open reading frame (ORF) sequence and therefore the DNA chips can be used for comparative genome hybridization (CGH) since the signals obtained are not sensitive to point changes and they average the information of the entire sequence [17]. Glass-slide microarrays and membrane-based macroarrays have both strengths and weaknesses. The probe density of microarrays is, by definition, higher than that of macroarrays. High density is, however, not a necessity in organisms such as yeast, with only 6000 genes. While membrane-based macroarrays are used in conjunction with radioactive labeling, glass microarrays can be used with fluorescent probes, which allows simultaneous use of a control reference and a sample in a two-color hybridization. Sensitivities are similar although radioactive labeling is more uniform than dual fluorescent labeling. Membrane arrays are easier to make and cheaper to use and thus offer an interesting alternative for many laboratories [1]. However, membrane arrays for yeast at affordable prices are not available. Moreover, the only existing ones have the probe dataset distributed into two membranes, making their use less convenient.
Our objective in this study was to develop an entire set of genes probes for the genome of S. cerevisiae and to use them in order to construct a new kind of macroarray, in which all the probes are included in an 8- to 11-cm single membrane. An additional advantage is that the cost of such macroarrays for the academic community is less than one third that of the commercial ones.
Materials and methods
Probe generation. Probes were obtained by three different methods:
(A) The general strategy was PCR amplification of each complete ORF inserted into a plasmid of the Research Genetics Exclone collection (Fig. 1A). This collection was originally developed by Eric Phyzicky's laboratory and contains 6080 clones. Each clone contains a different full-length yeast ORF inserted into a URA3 marker expression plasmid. [18]. Yeast cells were grown overnight at 30ºC in 96-well microtiter plates with 100 µl SC-URA medium: 0.17% yeast nitrogen base without amino acids (Difco), 0.5% ammonium sulfate, 2% glucose and 0.077% CSM-URA (complete supplement mixture without uracil, BIO 101). The cells were recovered by centrifugation, resuspended in 50 µl distilled water, frozen and stored for future uses.
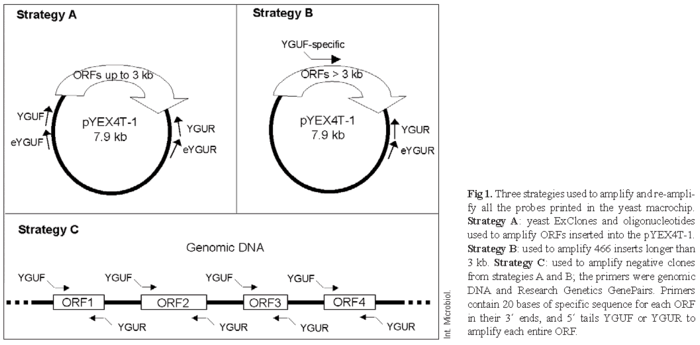
A first-round PCR was carried out in which the inserts were directly amplified from 8 µl of unfrozen cells using a pair of common primers (forward [eYGUF]: 5´-ATTCGATGATGAAGATACC-3´ and reverse [eYGUR] 5´-ACGATTCATAGATCTCTGC-3´) in a final volume of 100 µl. The reaction mix consisted of 1× Biotools buffer, 3 mM MgCl2, 0.2 mM of each nucleotide triphosphate, 0.5 µM of each primer and 4 U DNA polymerase (Biotools). Reactions were conducted in a thermocycler GeneAmp PCR System 9700 (Applied Biosystems) using the following protocol: 10 min at 94ºC to lyse the cells and denature the DNA, 30 cycles at 94ºC for 30 s, 50ºC for 30 s and 72ºC for 2-4 min (depending on the ORF length), followed by a final step of 72ºC for 10 min. In a second-round of PCR, 0.1-1 µl of the sample amplified in the first round was used for a second amplification with a pair of nested forward (YGUF) 5´-CGAATTCCAGCTGACCACCATG-3´ and reverse (YGUR) 5´-GATCCCCGGGAATTGCCATG-3´ general primers using the same Taq polymerase, PCR mix and thermocycler and the following steps: one cycle at 94ºC for 1 min, 30 cycles at 94ºC for 30 s, 60ºC for 30 s and 72ºC for 2-4 min (depending on the ORF length) and a final step of 72ºC for 10 min.
(B) A second strategy was used for genes longer than 3 kbp (Fig1B). An internal forward oligonucleotide compatible with eYGUR was designed using Pride software [10] or the Web primer interface at SGD (Saccharomyces Genome Database, http://www.yeastgenome.org/) at approximately 1000 bp upstream the ORF stop codon. Every oligonucleotide has a 5´ YGUF tail in order to allow a second round of amplification. PCRs were done as in strategy (A).
(C) Negative amplifications for genes shorter than 3 kbp, false positives from strategy A, and some ORFs included in the CYGD (Comprehensive Yeast Genome Database, http://mips.gsf.de/genre/proj/yeast/index.jsp, at July 2001) but not present in the Phyzicky's collection were PCR amplified using commercial GenePairs (Research Genetics) and genomic DNA as template (Fig. 1C) according to the following protocol: one cycle at 94ºC for 3 min, 30 cycles at 94ºC for 30 s, 50ºC for 30 s and 72ºC for 195 s and a final step at 72ºC for 10 min. A second round of amplification was performed as in strategy (A).
In all cases (three strategies and two PCR rounds), PCR products were analyzed on 0.8% agarose gels using 0.5× TBE as running buffer and DNA size markers. Some PCR products were quantified by fluorescent DNA assay in a Hoeffer DyNA Quant 200 Fluorometer (Hoeffer Pharmacia Biotech) according to the manufacturer's instructions. The sizes of the remaining samples were estimated by visual inspection of the ethidium-bromide-stained gels and comparison with standard markers of similar size.
Macroarray construction. BioGrid (BioRobotics, UK) was used as the spotting robot. Macroarrays were made by printing the PCR products (without purification) onto a positively charged nylon membrane (Amersham Hybond N+). Printing was done with a 384-pinhead printer, consisting of regular 4 × 4 spots per pin, yielding a total of 6144 available positions. Gene probes were printed in a single replicate. The chips contained positive control spots: 16 replicates of 0.2 ng total yeast genomic DNA and four replicates of a region of 18S rDNA. Negative control spots included four replicates of 0.2 ng Escherichia coli genomic DNA. Therefore, 6049 positions were used for gene probes, 24 for control probes and 71 remained empty.
Membranes were kept humid by setting them onto three Hybond blotting-paper sheets soaked in denaturing solution (1.5M NaCl, 0.5M NaOH). Solid pins of 0.4-mm diameter were used. These pins spot 20 nl each time. PCR products were spotted five times so that each spot contained 20-30 ng DNA in a diameter of approximately 0.6 mm. After printing, the membrane was neutralized with 1.5 M NaCl, 0.5 M Tris/HCl (pH 7.2), 1 mM EDTA (pH 8.0) for 1 min. No subsequent UV fixation was necessary (see Results) and membranes were kept on filter paper until complete dryness.
Microarrays on amino-coated glass slides were also constructed using purified PCR products. These will be described elsewhere (Viladevall et al., submitted).
Nucleic acid isolation. Genomic DNA was isolated from yeast cells by phenol extraction with glass beads, essentially as described by Hoffman and Winston [14]. Total RNA from yeast cells was prepared as described by Sherman et al. [23], but using a multiple-sample automated device (Fast-Prep, BIO101) to break the cells.
Radioactive sample labeling. Yeast genomic DNA was labeled following a random priming procedure. 800 ng genomic DNA in 45 µl distilled water was denatured for 5 minutes by boiling, added to a microcentrifuge tube of Ready-To-Go DNA labeling beads (Amersham Biosciences) and mixed with 50 µCi [α-33P] dCTP (≥2500 Ci/mmol). The mixture was incubated at 37°C for 1-2 h. In order to eliminate non-incorporated components, Probe Quant G-50 columns (Amersham Biosciences) were used.
Yeast cDNA was labeled by reverse transcription. The reaction mixture consisted of 30 µg total RNA, 0.5 µg oligo(dT) (5´-T15VN-3´), 1 µl RNAse OUT (Invitrogen), and RNA-treated water were added to a microcentrifuge tube in a total volume of 10 µl. The mixture was incubated at 70°C for 10 min and chilled on ice for 5 min. Five µl of 5× first-strand buffer, 3 µl 0.1M DDT, 1.5 µl dNTP mix (dATP, dGTP, dTTP 16 mM each, and dCTP 100 µM), 5 µl 33P-α-dCTP (10 µCi/µl) and 1 µl SuperScript II RT (200 U/µl, Invitrogen) were added in a final volume of 30 µl. The mixture was incubated at 43ºC for 1 h and the reaction stopped by adding 1 µl 0.5 M EDTA. Unincorporated nucleotides were removed by using MicroSpin S-300 HR columns (Amersham Bioscience) following the manufacturer's instructions.
Macroarray hybridization and stripping. Three different hybridization solutions were used: commercial Ambion ULTRArray™ hybridization buffer and SSC-based (5× SSC, 5× Denhart's, 0.5% SDS and 100 µg herring sperm DNA/ml) and SSPE-based (5× SSPE, 5× Denhart's, 50% deionized formamide, 0.5%, SDS, 10% dextran sulfate and 200 µg herring sperm DNA/ml) hybridization solutions. Different hybridization times, different amounts of radioactive sample and different exposure times were tested, as discussed below.
The hybridization protocol used was as follows. New macroarrays were pre-treated for 30 min at 80ºC with 0.5% SDS to remove particles deposited during array printing. This step was not necessary in subsequent hybridizations. Filters were inserted in 12.5 × 2.5-cm flat-bottom plastic tubes and pre-hybridized in a rotator oven with 5 ml pre-hybridization solution (the same as used for hybridization but without the radioactive sample) at the corresponding temperature for each solution (65ºC for SSC, 42ºC for SSPE and 50ºC for ULTRArray). The pre-hybridization solution was then replaced with 5 ml of the same solution containing the desired amount of radioactive sample and hybridized for the times described in Tables 1 and 2. For ULTRArray and SSC solutions, washing conditions were: 1× at 65ºC for 20 min in 2× SSC, 0.1% SDS, and twice at 65ºC for 30 min in 0.2× SSC, 0.1% SDS. For the SSPE solution, the washes were: twice at 42ºC for 10 min with 2× SSPE, SDS 0.1%, and once at 65ºC for 15 min with 1× SSPE, 0.1% SDS. After the washing step, membranes were kept humid, sealed in Saran wrap, avoiding any bubbles, and exposed to an imaging plate (BAS-MP, FujiFilm) for various times.
Filters were stripped by pouring 3×150 ml boiling stripping buffer (5 mM sodium phosphate, pH 7.5, 0.1% SDS) over the membrane. The first time, the stripping buffer was immediately changed while after the second and third washes the filters were left to cool at room temperature. To ensure that radioactivity had been eliminated, the filters were either checked with a Geiger counter or re-scanned with a Phosphorimager. Membranes were not dried at any time because drying can cause permanent fixation of the radioactivity. Once the radioactivity had been eliminated, membranes were stored dry for future use. We did not observe any decrease in macroarray performance during a period of 18 months after manufacturing.
Macroarray scanning and analysis. Images were acquired using a FujiFilm FLA3000 Phosphorimager. Spot intensities were measured as ARM density (artifact-removed density), background and sARM Density (background-corrected ARM density) by using the Array Vision software (Imaging Research, Canada).
p>Results and DiscussionProbe generation. The general strategy depicted in Fig. 1A was selected because the cost is about one third of that used for yeast PCR clones [8,11]. This strategy was followed at both laboratories of the authors and at the laboratory of Javier Arroyo at the Complutense University of Madrid (Spain). Of the 6080 clones, 4593 were amplified (success rate 75.5%). The fragment length that Taq polymerase can amplify under these conditions limited the yield. This strategy efficiently amplified only fragments shorter than 3 kbp. In Fig. 2, an example of a 72-set of second-round PCR products is given. Amplified bands that did not have the correct size were excised from the gel and sequenced. Most of them (e.g those marked in Fig. 2) corresponded to internal PCR sub-products or were multimers of the original sequence. Therefore, these reactions were considered positives, as they should not cause problems when used as a probe. In some cases, the extra bands had a sequence different from the expected one and were thus considered false positives. Negatives (arrow in Fig. 2) or false positives were subjected to strategy (C). An additional set of 5% of random selected clones was also sequenced and all of them had the correct sequence.

With strategy (B), 451 positive amplifications, out of 466 genes, were obtained, which represents a 96.8 % success rate. In addition to the ORFs amplified with commercial GenePairs (Research Genetics) and missing in the original Phyzicky's collection (strategy C), 56 new ORFs, discovered by global genome comparisons with other yeast species [3], were obtained by amplification using oligonucleotides designed by us and similar to the GenePairs. Most of this set of probes was produced in the laboratory of José L. Revuelta at the University of Salamanca (Spain). The global success of strategy (C), as judged by gel electrophoresis, was 95.2% (1029 out of 1080). The final yield of all the three strategies was 6073 probes.
Macrochip design. Efforts were made to improve other, previously described commercial or in-house yeast macroarrays [11]. A small size, 7 × 11 cm, was chosen in order to facilitate handling. This size requires less hybridization solution and less radioactive sample, and reduces the risk of breakage during manipulation.
The original yeast ORF collection was purged in order to eliminate ORFs that could give redundant information. Thus, some subtelomeric ORFs belonging to different gene families were eliminated (leaving one ORF member per gene family) to avoid cross-hybridization and therefore overlapping, non-useful information. In addition, clones corresponding to retrotransposon ORFs (Ty ORF) were also purged, leaving only one copy of each Ty ORF (Ty-1, Ty-2, Ty-3, Ty-4, Ty-5).
Finally, the probe collection was updated by comparing the ORF collection with the public yeast databases (i.e. SGD, CYGD). Thus, some ORFs turned out to be artifacts, for example, due to wrong assignations of the start/ending (some of them before re-sequencing of chromosome III), or complete or partial overlapping with other, well-reported genes, and were eliminated. After several trials in which some probes did not produce consistent results, 6049 of the 6073 probes amplified for the final version of the macrochip were selected: 451 from the 1020 bp amplified from the ORF 3´ end and 5518 that comprised the entire ORF.
The complete list of probes used in the macrochip and their grid locations can be obtained from the Web page of the authors: http://scsie.uv.es/chipsdna/. The DNA Chips platform has been registeredwith the accession number GPL772 at GEO database (http://www.ncbi.nlm.nih.gov/geo/).
Optimization of macroarray construction. Several parameters were tested in order to obtain the highest signal/background ratio. First, several positively charged commercial membranes were tried: Amersham Hybond N+, Schleider & Schuell Nitran supercharged, Boehringer Nylon membranes + charged, and Pall Biodyne B. Signals were compared by hybridizing the membranes with aliquots of the same radioactive genomic DNA sample. The best results (highest signal/noise ratio) were obtained using Amersham Hybond N+ or Pall Biodyne B (data not shown). In addition, 3, 5, 7 and 10 strokes of DNA samples from the first and second round PCR were tested. The concentration of DNA from the first round of PCR (2-5 ng/µl) was not enough to yield a good signal even after 10 strokes. Instead, 5 strokes with a DNA concentration of about 200 ng/µl, obtained by second-round PCR, was determined to be the optimum condition (data not shown).
The influence of membrane humidity on the shape of the spot was also evaluated. If the membrane was not humid enough, the spots had a doughnut shape; if the membrane was too humid the spots had irregular borders. To achieve well-defined round spots, liquid in excess was drained from the tray system and the surface of the membranes was allowed dry for a few minutes before printing.
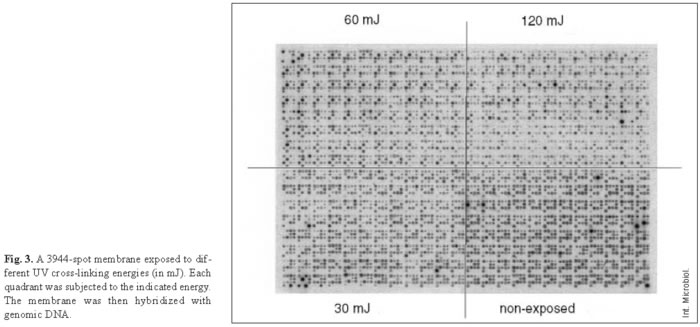
Usually, macroarray protocols include, at the end of each printing process, a DNA fixation step with UV cross-linking [11]. In order to determine the optimum energy of cross-linking, the same membrane was divided into four sections using masks. Three parts were exposed, respectively, to 125, 60, 30 mJ of UV light and one part was not exposed (Fig. 3). The results indicated that the best method is no UV irradiation, probably because the alkali printing itself constitutes a good fixation method. It may also be that UV treatment damages the fixed DNA and subsequently the signal decreases as UV energy increases [21]. The non-UV-treated part of the filter behaved better than the UV-treated portions even after the same membrane was reused eight times (not shown).
Evaluation of the hybridization conditions. DNA macrochips are not as widely used as microchips. For this reason, the protocols for their use, although similar to those used for other routine molecular-biology experiments, need to be optimized. Hybridization conditions were optimized in order to obtain the best possible results with respect to signal/noise ratio after each use of the membranes and to extend the number of uses as much as possible. The experiment shown in Table 1A was designed to determine the best hybridization solution. It can be seen that the commercial Ambion ULTRArray hybridization buffer gave the highest total signal (the sum of all the individual spot signals minus the background, represented by ΣsARM density) whereas the lowest signal was obtained using SSC solution. A possible reason for the better performance of the ULTRArray and SSPE solutions is that they reduce the time required for final equilibrium conditions. Both solutions contain formamide as an accelerator (although the composition of ULTRArray is not given by the supplier, commercial rapid-hybridization buffers usually contain formamide and cationic detergents or dextran sulfate) [21]. Nevertheless, the effect of both formamide and buffer composition on hybridization rate is controversial [5]. In our hands, SSPE and, especially, ULTRArray solutions shortened the time required for a reasonably good result. In order to achieve the same signal intensity with the SSC solution, longer hybridization times would be needed. The increase in total hybridization signal seen in experiment 2 after 41 h compared with 17 h in experiment 1 supports this hypothesis. When ULTRArray solution is used, shortening the hybridization time (Table 1B, compare experiments 7 and 9) to 5 h, it decreases the signal to a level similar to that obtained in SSC solution after a 17-h hybridization. Hybridization conditions were evaluated further by comparing the exposure times and radioactive sample concentrations. The results show that the signal was linearly by a factor of three when the exposure time is reduced from 17 to 5 h (Table 1B, experiments 5 vs. 6, 7 vs. 8, 9 vs. 10). The concentration of radioactive sample (in dpm/ml of hybridization solution) is also important. Doubling the amount produces approximately double the signal (experiment 5 vs. 7) within the range of our experiments (1 to 5 × 106 dpm/ml). The increase in signal intensity is not, however, the only factor to be considered. It can be seen in Table 1A, B that the signal/background ratio is better when higher sample concentrations are used (compare experiments 1-4 with experiments 5-6 and 7-10) and is mostly independent of the hybridization solution. The higher the average ratio obtained, the higher the number of valid spots observed (valid spots are usually those with ratios 1.5- to 2-fold over the background) [4]. We have observed, however, that in cDNA hybridizations SSPE and ULTRArray tend to produce higher and irregular backgrounds (not shown). The high background in formamide-containing solutions was also observed by other authors [13]. Thus, the SSC solution is the best option for hybridization of macroarrays. Moreover, the use of longer hybridization times in SSC solution does not significantly bias the results (compare experiments 1 vs. 2, Tables 1A and 2).
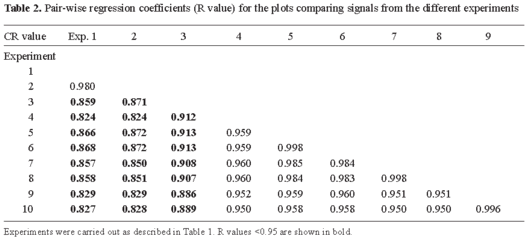
The hybridization solution should be chosen with caution because of potential differences in probe-target specificity [5]. To test the influence of different hybridization solutions, hybridization times and sample concentrations on the individual probe signals, a set of pair-wise comparisons was made between the complete set of spot intensities (Table 2) and the R (Pearson coefficient) was estimated in each case. The R value was very good (>0.95) in two of the comparisons: when the same hybridization solution was used and, especially, when the same membrane was used. While the decrease in the R values of different filters was expected, the small effect (0.02-0.04 R units) indicates that the macroarrays that we produced are very uniform and yield reproducible results. In fact, the effect of using samples labeled independently was higher than the effect of using different membranes (not shown). Nevertheless, we recommend using the same membrane for comparative experiments. R values were below 0.95 when different hybridization solutions were compared (the same labeled reaction sample was used for experiments 1-4 and another one for experiments 5-10, both divided into aliquots). This can be explained by considering that each hybridization solution may have different efficiencies for sequences with base composition outside the G + C range of 30-75% [21]. The use of the same hybridization solution in comparative experiments is, therefore, mandatory. Finally, a comparison among R values of different solutions showed that SSPE and ULTRArray behave similarly compared to SSC solution.
To summarize, the best results were obtained with SSC solution, about 5 × 106 dpm/ml of genomic DNA radioactive sample (2-3 × 106 dpm/ml if using cDNA instead, not shown) and a hybridization time of 40 h; under these conditions, both the highest number of valid spots and the best signal/noise ratio are obtained. However, other hybridization solutions, sample concentrations and hybridization times can also be used in combination with appropriate exposure times to produce fairly good results with our membrane macroarrays.
Number of uses of the same chip. Studies involving DNA chips are always done by means of comparative experiments. When working with radioactivity, two hybridization steps, at least, with a stripping step in between, are needed [4]. The reusing of macroarrays is, then, strictly necessary. Therefore, it is important to know how many times the same membrane can be reused. In a study with a simplified version of the macrochips described here (3944 spots instead of 6050), the same membrane was hybridized and subsequently stripped nine times. Spots with a signal higher than 1.5 times the background were considered valid. The number of valid spots in each experiment was calculated as a percentage based on the total number of spots (Fig. 4A). The results showed that there was no significant decrease in the number of valid spots after the second use and an average decrease of only 3% compared with the first use was observed throughout the entire series. These findings are similar to those reported by Hauser et al. [11]. After nine uses, 97% of the spots still produced valid results and the background was similar to that of the first hybridization (Fig. 4B). In other experiments, the same membrane was used up to 12 times with satisfactory results (data not shown).

Applications of the study. The macroarrays described here had already been used in several different studies and proved to be both highly convenient and reliable [2,9,19,27]. Their small size and high-quality printing allow them to be hybridized many times, which not only extends the use of the yeast macroarrays but also greatly reduces the cost of the experiments. Since no other DNA chip developed to date has been used so many times [1], they allow comparison of many different samples in the same chip, which improves the statistical significance of the experiment.
The probe collection set generated has other applications as well. Because all of the probes, independently of the protocol used for their synthesis (strategies A-C), have common sequences at their ends (YGUF/YGUR), they can be re-amplified either as a complete set or individually using the same PCR conditions (second-round PCR described in Materials and methods). This has been done in our laboratories for the generation of probes for Northern or Southern experiments and for making specialized "minichips" for specific purposes, such as for a study of chromosome I polymorphisms [6].
Acknowledgements. This work was supported by grants 1FD97-1254-C02-01 from the Ministry of Science and Technology (Spain), AE2001-100 from the Generalitat Valenciana (Spain) and QLRI-CT-1999-01333 from the EU to J.E.P.O; BIO2001-4357-E from the Ministry of Science and Technology (Spain) and 2000ACES 00041 (Autonomous Government of Catalonia) to J.A. O.A. has a fellowship from the Ministry of Education and Culture (Spain).
References
1. Becker KG, Wood WH, Cheadle C (2002) Membrane-based spotted cDNA arrays. In: Bowtell D, Sambrook J (eds) DNA microarrays. A molecular cloning manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp. 289-306 [ Links ]
2. Bellí G, Molina MM, García-Martinez J, Pérez-Ortín JE, Herrero E (2004) Saccharomyces cerevisiae glutaredoxin 5-deficient cells subjected to continuous oxidizing conditions are affected in the expression of specific sets of genes. J Biol Chem 279:12386-12395 [ Links ]
3. Blandin G, Durrens P, Tekaia F, Aigle M, Bolotin-Fukuhara M, Bon E, Casaregola S, de Montigny J, Gaillardin C, Lepingle A, Llorente B, Malpertuy A, Neuveglise C, Ozier-Kalogeropoulos O, Perrin A, Potier S, Souciet J, Talla E, Toffano-Nioche C, Wesolowski-Louvel M, Marck C, Dujon B (2000) Genomic exploration of the hemiascomycetous yeast: 4. The genome of Saccharomyces cerevisiae revisited. FEBS Letters 487:31-36 [ Links ]
4. Bowtell D, Sambrook J (2002) DNA microarrays. A molecular cloning manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [ Links ]
5. Brown T (1995) Hybridization analysis of DNA blots. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Current Protocols in Molecular Biology, vol. 1. John Wiley, New York, pp. 2.10.1-2.10.16 [ Links ]
6. Carro D, García-Martínez J, Pérez-Ortín JE, Piña B (2003) Structural characterization of chromosome I size variants from a natural yeast strain. Yeast 20:171-183 [ Links ]
7. Cox KH, Pinchack AB, Cooper TG (1999) Genome-wide transcriptional analysis in S. cerevisiae by mini-array membrane hybridization. Yeast 15:703-713 [ Links ]
8. DeRisi JL, Iyer VR, Brown PO (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680-686 [ Links ]
9. García-Martínez J, Aranda A, Pérez-Ortín JE (2004) Genomic Run-On evaluates transcription rates for all yeast genes and identifies new regulatory mechanisms. Mol Cell 15:303-313 [ Links ]
10. Haas S, Vingron M, Poustka A, Wiemann S (1998) Primer design for large scale sequencing. Nucleic Acids Res 26:3006-3012 [ Links ]
11. Hauser N, Vingron M, Scheideler M, Krems B, Hellmuth K, Entian K-D, Hoheisel J (1998) Transcriptional profiling on all open reading frames of Saccharomyces cerevisiae. Yeast 14:1209-1221 [ Links ]
12. Hauser NC, Fellenberg K, Gil R, Bastuck S, Hoheisel JD, Pérez-Ortín JE (2001) Whole genome analysis of a wine yeast strain. Comp Funct Genom 2:69-79 [ Links ]
13. Hayes A, Zhang N, Wu J, Butler PR, Hauser NC, Hoheisel JD, Lim FL, Sharrocks AD, Oliver SG (2002) Hybridization array technology coupled with chemostat culture: tools to interrogate gene expression in Saccharomyces cerevisiae. Methods 26:281-290 [ Links ]
14. Hoffman CS, Winston F (1987) A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57:267-272 [ Links ]
15. Holstege, FCP, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95:717-728 [ Links ]
16. Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, Kobayashi S, Davis C, Dai H, He YD, Stephaniants SB, Cavet G, Walker WL, West A, Coffey E, Shoemaker DD, Stoughton R, Blanchard AP, Friend SH, Linsley PS (2001) Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol 19: 342-347 [ Links ]
17. Infante JJ, Dombek KM, Rebordinos L, Cantoral JM, Young ET (2003) Genome-wide amplifications caused by chromosomal rearrangements play a major role in the adaptive evolution of natural yeast. Genetics 165:1745-1759 [ Links ]
18. Martzen MR, McCraith SM, Spinelli SL, Torres FM, Fields S, Grayhack EJ, Phizicky EM (1999) A biochemical genomics approach for identifying genes by the activity of their products. Science 286:1153-1155 [ Links ]
19. Rodríguez-Navarro S, Fischer T, Luo M-J, Antúnez O, Brettschneider S, Lechner J, Pérez-Ortín JE, Reed R, Hurt E (2004) Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell 116:75-86 [ Links ]
20. Posas F, Chambers JR, Heyman JA, Hoeffler JP, de Nadal E, Ariño J (2000) The transcriptional response of yeast to saline stress. J Biol Chem 275:17249-17255 [ Links ]
21. Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [ Links ]
22. Schena M, Shalon D, Davis RW Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467-470 [ Links ]
23. Sherman F, Fink GR, Hicks JB (1986) Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [ Links ]
24. Talla E, Tekaia F, Brino L, Dujon B (2003) A novel design of whole-genome microarray probes for Saccharomyces cerevisiae which minimizes cross-hybridization. BMC Genomics 4:38-56 [ Links ]
25. Wodicka L, Dong H, Mittmann M, Ho MH, Lockhart DJ (1997) Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotech 15:1359-1367 [ Links ]
26. Zuzuarregui A, del Olmo M (2004) Expression of stress response genes in wine strains with different fermentative behavior. FEMS Yeast Res 4:699-710 [ Links ]

