










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
versión impresa ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.9 no.5 nov./dic. 2004
Tooth agenesis: in search of mutations behind failed dental development
KOLENC-FUSÉ FJ. TOOTH AGENESIS: IN SEARCH OF MUTATIONS BEHIND FAILED DENTAL DEVELOPMENT. MED ORAL PATOL ORAL CIR BUCAL 2004;9:385-95.
ABSTRACT
Tooth agenesis are the most common craniofacial malformations. Its prevalence in permanent dentition reaches 20% and its expressivity ranges from only one tooth, usually a third molar, to the whole dentition. Genetic linkage and molecular biology studies have allowed, in the last decade, the identification of mutations responsible for some patterns of syndromic and non-syndromic tooth agenesis. The mutated genes are key genes for the development of dentition, like the ones that encode the transcription factors MSX1, PAX9 and PITX2, the signalling protein EDA and its receptor EDAR. Current research would lead to the development of new classifications of tooth agenesis that took into account both the phenotypes and the genetic background. This would allow an early diagnosis of the condition, before the development of the somatic defect, that could eventually be repaired with gene therapy or tissue and organ engineering.
Key words: Tooth agenesis, hypodontia, oligodontia, PAX9, MSX1.
INTRODUCTION
Mammalian dentition is a segmented system, composed of a series of homologous elements, sharing a similar structure but different in shape and size (1). It is analogous to the vertebral column in that a modular structure repeats itself with modifications to constitute a complex system. In this kind of system, some units can be absent due to lack of development, and that is what is called an agenesis. Tooth development is a complex process, in which reciprocal and sequential interactions between epithelial and mesenchymal cells (2) regulate cell activities like proliferation, condensation, adhesion, migration, differentiation and secretion, that lead to the formation of a functional tooth organ. Organogenesis involves three fundamental processes: a) initiation, in which a group of cells interprets positional information provided by other cells to initiate organ formation at both the right place and time, b) morphogenesis, in which the cells build up an organ rudiment and c) differentiation, in which cells build up organ-specific structures (3). Recent advances in our understanding of molecular aspects of odontogenesis (2-5) indicate that the development of teeth is under strict genetic control, which determines the positions, numbers and shapes of different types of teeth (6). Most studies have been done using mice as models. The scarce direct knowledge on the molecular basis of human odontogenesis derives from the study of pathological cases. More than 200 genes are known by now to have a role in odontogenesis (7). Proteins coded by these genes have many functions, being transcription factors, signalling molecules and its receptors and extracellular matrix molecules some of the more relevant for tooth development. Mutations in any of these genes could eventually lead to failures in odontogenesis. The earlier these proteins are needed for the development, the worse the malformation derived from their altered function may be. An alteration in the function of a protein needed for the initiation or early morphogenesis could lead to agenesis. Many proteins can have different functions during the various processes of organogenesis, during the development of the different kinds of teeth or during the development of primary and permanent dentitions. This could explain the association between different dental anomalies like agenesis and delayed eruption and alterations in shape, size and position of the other teeth (6, 8-11).
Agenesis of at least one tooth is the most common anomaly of dental development (12). Lack of up to five teeth is called hypodontia; lack of six or more teeth is called oligodontia (13), and anodontia is the failure of development of the whole dentition. The prevalence of permanent tooth agenesis ranges between 1.6% and 9.6%, depending on the studied population and it reaches 20% if third molars are considered (12). The prevalence in primary dentition is lower, ranging between 0.5% and 0.9% (12). The most common missing teeth are third molars, followed by lateral maxillary incisors or second mandibular premolars (12).
Tooth agenesis can be isolated, as the only phenotypic alteration in a person, or associated to other alterations as part of a syndrome. Isolated, non-syndromic agenesis can be sporadic or familiar; and may be inherited in mendelian dominant or recessive autosomic mode, or X-linked (12). Penetrance has been traditionally considered as incomplete but high. The expressivity of the various forms is quite variable, with a wide range of missing teeth, despite the fact that a typical phenotype can be defined for each known genetic defect. Some researchers consider the alterations in shape, like mesio-distally reduced or peg-shaped teeth, as a variation in the expression of the mutated genes. Effects of modifying genes (14) or epigenetic factors could explain this variation. It should be taken into account that the activity of these molecules is regulated by the interaction with other molecules that can be tissue-specific and have allelic variants, and the interplay between them could result in different phenotypic outcomes. Many of the genes with a role in tooth development also have important functions in the development of other organs. This explains the finding of tooth agenesis in at least 45 syndromes (15)
NON-SYNDROMIC TOOTH AGENESIS
Molar oligodontia. MIM 604625.
This form of autosomal dominant oligodontia is characterized by agenesis of most permanent molars, and can eventually involve other teeth like second premolars and central mandibular incisors (16-19). Figures 1 and 2 summarize the phenotype of published cases. Primary molars can be missing in the more severe cases (17, 19). The existing teeth can show a mesio-distal reduction in size, or be peg-shaped incisors. Many mutations in the PAX9 gene, mapped to 14q12-q13, were found in patients affected by this form of oligodontia (Table 1). The mutations would lead to a loss of the protein function, and haploinsufficiency would be the pathogenic mechanism (17, 19). The most severe phenotype published to date is due to a heterozygous deletion of PAX9 locus (13), supporting the haploinsufficiency mechanism proposed, and indicating that in the other mutations, like the missense or nonsense ones, some biological activity of the proteins could remain.

PAX9 belongs to a transcription factor family (20) with nine members in mammals, characterized by a DNA-binding domain called "paired domain" and an additional homeodomain in most members (but not in PAX9). They are important regulators of organogenesis that can trigger cellular differentiation or maintain the pluripotency of cell populations in development. The functions of PAX9 in development have been studied in mice (21). Pax9 is widely expressed in the neural crest-derived mesenchyme involved in craniofacial and tooth development. Pax9 -/- mice show cleft secondary palate -besides other skeletal alterations-, lack thymus and parathyroid glands and all teeth are absent. Tooth development is arrested at the bud stage. At this stage Pax9 is required for the mesenchymal expression of Bmp4, Msx1 and Lef1, suggesting that its function is essential to establish the inductive capacity of this tissue (21). Nevertheless, Pax9 +/- mice are normal. Taken together, these findings suggest that PAX9 is a dosage-sensitive gene in humans, and that somehow its function is more important in the posterior teeth, especially in those derived from the proliferation of the dental lamina that gives origin to the permanent molars. Research in primates is needed to clarify the expression and function of this gene in a bifiodontic dentition.
Second premolar and third molar hypodontia. MIM 106600.
Vastardis's work fructified in 1996 with the identification of the underlying genetic cause for this form of dominantly-autosomic inherited non-syndromic hypodontia (22). It is characterized by second premolars and third molars agenesis, involving also other teeth. In a Dutch family, three of the 11 affected members had also cleft palate (23). In another family (24), existing teeth were smaller, second upper molars lacked the distal lingual cusp and mandibular first molars lacked the distal buccal cusp. Phenotype of published cases is summarized in figures 3 and 4. The responsible mutations (Table 2) were found in the MSX1 gene, mapped to 4p16.1. The expression of this gene is observed very early in odontogenic mesenchyme (7).
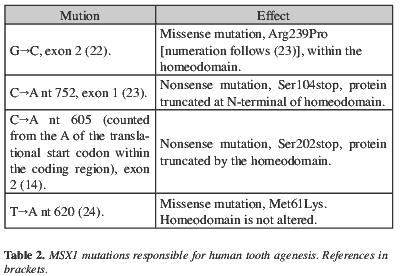
Msx1-/- mice have cleft secondary palate, lack all teeth - whose development is arrested at bud stage- and have skull, jaw and middle ear defects (25). MSX genes encode a group of homeodomain transcription factors required in different stages of development, like patterning, morphogenesis and histogenesis, and they function as transcriptional repressors (25). They are expressed in undifferentiated multipotential cells that are proliferating or dying; provide positional information and regulate epithelial-mesenchymal signalling in craniofacial development (25). It has been shown that MSX1 inhibits cell differentiation by maintaining high levels of cyclin D1 expression and Cdk4 activity, thus preventing the exit from the cell cycle and keeping the cells able to respond to proliferative factors (26). The loss-of-function mutations would lead these cells to differentiate earlier and stop proliferating, producing impaired morphogenesis (26). The mutation identified by Vastardis et al. (22) involves the substitution of a key aminoacid residue within the homeodomain and leads to a less stable protein, with little or no ability to bind DNA and lacking biological activity (27); accordingly, the phenotype is due to haploinsufficiency. A mutation found outside the homeodomain (24) remarks the role of the N-terminal region for the interaction of MSX1 with other proteins needed for transcription. The selective effect on specific types of teeth can be explained by a different sensitivity to the protein concentration in the tissues of the different developing types of teeth (27); by the timing of expression of MSX1; in combination with the presence or absence of functional redundancy from other genes during tooth morphogenesis (14); or by different genetic mechanisms for the development of the different tooth types in primates.
He-Zhao deficiency. MIM 604625.
This condition was characterized in a kindred from northwest China, in which 52 out of 328 members, belonging to six generations, were affected (28). The mode of inheritance is autosomal dominant, with an estimated penetrance of 88% and very variable expressivity. Primary dentition is not affected. The number of absent permanent teeth is very variable, eventually reaching the whole dentition, being the most commonly missing teeth the third molars, second premolars and lateral upper incisors. The disease locus has been mapped to a 5.5 cM region on 10q11.2 (29). Several genes in this region can be considered as candidates, such as Dkk-1, that codes a protein which is an antagonist to Wnt signalling; PRKG1B, that produces a cGMP-dependent protein kinase; and a KOX zinc finger gene cluster (29). Researches are currently conducted to detect mutations in this genes.
Incisor-premolar hypodontia (IPH). MIM 150400.
This is the most common form of inherited hypodontia. A Finnish research team has been studying its genetic basis for a decade (6, 30). The mean number of missing teeth in probands is 2.3. The most frequently teeth missing are lower second premolars (47%), upper second premolars (30%), upper lateral incisors (17%), and lower central incisors (4.2%) -third molars were excluded from the study-. Primary dentition is not affected. Calculated penetrance is 97%, and it is transmitted in an autosomal dominant manner. The authors considered peg-shaped teeth as a variation in expression. Many dental anomalies are associated with this form of hypodontia: ectopic palatally displaced upper canines, rotated premolars and taurodontism (6). The genetic cause for this condition has not been found yet, but mutations in MSX1, MSX2, EGF, EGFR and FGF-3 -all of them with roles in dental development (7)- have been excluded (30, 31).
Autosomal recessive hypodontia. MIM 602639.
Ahmad et al. (32) characterized this condition of variable expressivity from a highly consanguineous family from Pakistan. Hypodontia is associated with incomplete development of almost all teeth, malformation of crowns, lack of root development, enamel hypoplasia and failure of eruption. The disease locus has been mapped to a yet unidentified gene in chromosome 16q12.1.
Recessively inherited lower incisor hypodontia (RIH).
Pirinen et al. (11) described this condition in patients from 34 Finnish families. It is characterized by the absence of several lower incisors and upper lateral permanent incisors, also involving other teeth, particularly second premolars. In half of the patients, the corresponding deciduous teeth had either been missing or peg shaped. It is associated with other anomalies, like taurodontism, eruption delays and atopic conditions. The authors believe this condition can be the same formerly reported by others from various countries. Its high prevalence in Finland may be explained by the genetic isolation of the population of this country, and RIH can belong to the Finnish Disease Heritage, a collection of some 40 rare disorders, many of them recessive.
SYNDROMIC TOOTH AGENESIS
Tooth agenesis is commonly associated with other abnormalities in many syndromes, because many genes take part in common molecular mechanisms between tooth and other organs' development. We will now describe the recent findings into the molecular basis of three of these syndromes.
Anhydrotic ectodermal dysplasia (EDA). MIM 305100.
Ectodermal dysplasias are a group of some 150 conditions that involve anomalies in at least two of the following ectodermal-derived structures: hair, skin, nails, and teeth (33). Anhydrotic ectodermal dysplasia is characterized by hypohidrosis, hypotrichosis and hypodontia. The absence of most primary and permanent teeth is a characteristic finding. Present teeth usually have a conical crown shape. Taurodontism can be present. This syndrome has X-linked inheritance, so it occurs mostly in males, although heterozygous women may express the condition (34). The disease is produced by point mutations, deletions or translocations in the ED1 (=EDA) gene, mapped to Xq12-q13.1 (35, 36). ED1 encodes ectodysplasin-A, a 391 aminoacid protein, that belongs to the TNF-ligand family. It has a cytoplasmic, a transmembrane and an extracellular domain. Within the last domain are: a region with a repetitive collagen-like sequence Gly-x-y needed for the trimerization of the protein; various sequences highly conserved in TNF family; and a site for furin proteolitic cleavage (36). This cleavage would allow the protein to act as a diffusible signal. ED1 is expressed in the developing hair follicles, sweat glands and teeth, and it plays an important role in epithelial-mesenchymal signalling. Mutations have been found in DL (=EDAR), mapped to 2q11-q13, the gene that codes the ED1 receptor, that produces recessive or dominant autosomic forms of EDA whose phenotype is indistinguishable from the X-linked form (37). EDA/EDAR signalling activates transcription factor NF-kappaB through IKK (IkappaB kinase) complex (38). In mice, Ta (the homologue of ED1) is expressed in the oral epithelium and in the outer enamel epithelium during tooth development; while Dl (the homologue of DL) is expressed first in the oral epithelium and then it is restricted to the enamel knot -a signalling center that controls proliferation and apoptosis, and regulates cusp development- (39). In mice with mutations in Ta, the enamel knot is smaller and the expression of the marker genes needed for signalling (Shh, Fgf-4, Bmp-4 and Wnt10b) is weak (39). Instead, mutations in Dl lead to failure in enamel knot development, that appears as an elongated band of disorganized cells, though signalling activity is retained (39). These facts remark the role of the EDA/EDAR signalling pathway in enamel knot organization as a signalling center, and its function in tooth morphogenesis, especially in the cusp patterning.
Witkop "tooth and nail" syndrome. MIM 189500.
This syndrome is also an ectodermal dysplasia. It is characterized by hypo or oligodontia and nail dysgenesis. The existing teeth may be cone-shaped, with short roots or taurodontiform molars. Primary dentition can be affected. The mode of inheritance is autosomal dominant. Nails are hypoplastic, spoon-shaped, being the toenails more severely affected than fingernails (14, 34). The expressivity is quite variable. A nonsense mutation within MSX1 homeobox (4p16.1) has been identified as responsible for this disorder (14). The protein produced from the mutated allele would be truncated, and lacking the entire C-terminal region and the II and III homeodomain helixes, that are important for protein stability and DNA binding. The mutant protein would have no biological function, and haploinsufficiency is probably the pathogenic mechanism (14). Msx1 mesenchymal expression is important for both normal nail and tooth development in mice (14, 40); this data suggests that this transcription factor shares a common function or mechanism in the development of those structures. The tooth phenotype is more severe in this syndrome than in other non-syndromic tooth agenesis produced also by MSX1 mutations (figs. 3 and 4), although the pattern of missing teeth is coincident and shows the selectivity of the MSX1 function in human odontogenesis.
Rieger syndrome Type 1. MIM 180500.
It is characterized by hypodontia, malformation of the anterior chamber of the eyes and umbilical anomalies. There is midfacial hypoplasia and underdevelopment of the premaxillary area. The upper maxillary deciduous and permanent incisors and second upper premolars are most commonly missing. The lower anterior teeth have usually conical crowns, and cleft palate may be present. The mode of inheritance is autosomal dominant, with almost complete penetrance and variable expressivity (34). Mutations responsible for this malformation have been found in PITX2 (=RIEG), a gene mapped to 4q25-q26, that encodes a bicoid-related homeodomain transcription factor (41). Pitx2 expression is one of the first markers of tooth development at the initiation stage (2), previous to any morphological manifestation. It is expressed in the odontogenic areas of oral epithelium, in the periocular mesenchyma and the umbilicus (41). Wnt signalling induces PITX2 expression. PITX2 promotes cell proliferation by regulating the expression of genes that control cell cycle at G1 stage, like cyclin D2; and provides a link between Wnt and growth factor pathways in synergistically mediating cell type-specific regulation of growth control gene expression (42). PITX2 also upregulates DLX2 expression - another transcription factor with a role in tooth development - by binding to its promoter (43). The mutations responsible for this syndrome would be loss-of-function ones, supporting haploinsufficiency as the proposed pathogenic mechanism (43).
CONCLUSIONS
The discovery of genes that take part in tooth development programs and the identification of mutations responsible for craniofacial malformations, allow us to start understanding the ethiology and pathogenic mechanisms of these conditions. Tooth agenesis is a complex trait, with variable expressivity, and it appears associated with other tooth alterations. Mutations found by now can explain only a little percentage of the observed prevalence (44). The fact that mutations in a single gene, like MSX1, produce either an isolated or a syndromic -as extension of the phenotype - form of tooth agenesis, supports the assumption that the discovery of mutations responsible for syndromes that have tooth abnormalities, could be a tool to identify genes whose mutations explain the isolated forms (44).
The finding that mutations in certain genes selectively affect the development of only some kind of teeth, supports the conclusions of the studies made with genetically altered mice, that there would be different underlying genetic mechanisms for the different kind of teeth. They also support the existence of the odontogenic homeobox code proposed by Sharpe (45), equivalent to the one that determines the identity of the limb bones (46).
New techniques are already available for the early diagnosis of mutations that imply a risk of developing genetic diseases. The combination of genetic and clinic studies could lead to satisfactory classifications of these anomalies, that combine the phenotypes with the underlying genetic defects. This would bring new possibilities of early diagnosis and the foresight of orthodontic, surgical or prosthetic treatment. Recent advances in tissue and organ engineering and gene therapy, could even allow the implantation of cultured tooth germs or the early repair of the genetic defect, leading to normal development (47).
The recording of cases by clinicians is of great importance for future research. The proper study of familial hereditary and sporadic forms of tooth agenesis is essential for the discovery of new gene mutations that cause these anomalies. When a case of tooth agenesis is seen, the presence of the anomaly should be investigated in the other family members; and the case should be recorded with a complete clinical history including panoramic radiographs and models, for proper phenotype characterization prior to any surgical or orthodontic treatment. A precise description of missing teeth and any other tooth malformation -of size, shape, position, eruption or even structural- is needed, as well as the search for alterations in other organs and systems. Current methods allow taking DNA samples by non-invasive methods like a swab of the oral mucosa, with minimum discomfort for the patient. The conjunction of clinical and molecular studies is the right way to improve our understanding of the causes of these alterations.
AKNOWLEDGEMENTS
The author wants to thank Mariana Juambeltz, Claudio Martínez Debat and Enrique Zinemanas who reviewed the various versions of the manuscript and made valuable suggestions and corrections; Natalia Acevedo and Graciela Duarte made suggestions that improved the text; Clare Rymer for her support in the library of the Faculty of Dentistry/UDELAR; Elizabeth Lettier for the correction of the English version of the text; and a lot of compatriots scattered around the world who supplied valuable papers that made possible this review.
REFERENCES
1. Stock DW, Weiss KM, Zhao Z. Patterning the mammalian dentition in development and evolution. BioEssays 1997;19:481-90. [ Links ]
2. Jernvall J, Thesleff I. Reiterative signalling and patterning during mammalian tooth morphogenesis. Mech Dev 2000;92:19-29. [ Links ]
3. Peters H, Balling R. Teeth. Where and how to make them. Trends Genet 1999; 15:59-65. [ Links ]
4. Cobourne MT. The genetic control of early odontogenesis. Br J Orthodontics 1999;26:21-8. [ Links ]
5. Tucker AS, Sharpe PT. Molecular genetics of tooth morphogenesis and patterning: the right shape in the right place. J Dent Res 1999;78:826-34. [ Links ]
6. Arte S, Nieminen P, Apajalahti S, Haavikko K, Thesleff I, Pirinen S. Characteristics of incisor-premolar hypodontia in families. J Dent Res 2001;80:1445-50. [ Links ]
7. Gene Expression in Tooth. http://bite-it.helsinki.fi/ [ Links ]
8. Apajalahti S, Arte S, Pirinen S. Short root anomaly in families and its association with other dental anomalies. Eur J Oral Sci 1999;107:97-101. [ Links ]
9. Goldenberg M, Das P, Messersmith M, Stockton DW, Patel PI, D'Souza RN. Clinical, radiographic and genetic evaluation of a novel form of autosomal-dominant oligodontia. J Dent Res 2000;79:1469-75. [ Links ]
10. Pirinen S, Arte S, Apajalahti S. Palatal displacement of canine is genetic and related to congenital abscence of teeth. J Dent Res 1996;75:1346-52. [ Links ]
11. Pirinen S, Kentala A, Nieminen P, Varilo T, Thesleff I, Arte S. Recessively inherited lower incisor hypodontia. J Med Genet 2001;38:551-6. [ Links ]
12. Vastardis H. The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop 2000;117:650-6. [ Links ]
13. Das P, Stockton DW, Bauer C, Shaffer LG, D'Souza RN, Wright JT, et al. Haploinsufficiency of PAX9 is associated with autosomal dominant hypodontia. Hum Genet 2002;110:371-6. [ Links ]
14. Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, Seidman CE, et al. A nonsense mutation in MSX1 causes Witkop Syndrome. Am J Hum Genet 2001;9:743-6. [ Links ]
15. On Line Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/Omim/ [ Links ]
16. Stockton DW, Das P, Goldenberg M, D'Souza RN, Patel PI. Mutation of PAX9 is associated with oligodontia. Nat Genet 2000;24:18-9. [ Links ]
17. Nieminen P, Arte S, Tanner D, Paulin L, Alaluusua S, Thesleff I, et al. Identification of a nonsense mutation in the PAX9 gene in molar oligodontia. Eur J Hum Genet 2001;9:743-6. [ Links ]
18. Frazier-Bowers SA, Guo DC, Cavender A, Xue L, Evans B, King T, et al. A novel mutation in human PAX9 causes molar oligodontia. J Dent Res 2002; 81:129-33. [ Links ]
19. Das P, Hai M, Elcock C, Leal S, Brown D, Brook A, et al. Novel missense mutations and a 288-bp exonic insertion in PAX9 in families with autosomal dominant hypodontia. Am J Med Genet 2003;118:35-42. [ Links ]
20. Chi N, Epstein JA. Getting your Pax straight: Pax proteins in development and disease. Trends Genet 2002;18:41-7. [ Links ]
21. Peters H, Neubuser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 1998;12:2735-47. [ Links ]
22. Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 1996;13:417-21. [ Links ]
23. van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HKP. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 2000;24:342-3. [ Links ]
24. Lidral AC, Reising BC. The role of MSX1 in human tooth agenesis. J Dent Res 2002;81:274-8. [ Links ]
25. Bendall AJ, Abate-Shen C. Roles for Msx and Dlx homeoproteins in vertebrate development. Gene 2000; 247:17-31. [ Links ]
26. Hu G, Lee H, Price SM, Shen MM & Abate-Shen C. Msx homeobox genes inhibit differentiation through upregulation of cyclin D1. Development 2001; 128:2373-84. [ Links ]
27. Hu G, Vastardis H, Bendall AJ, Wang Z, Logan M, Zhang H, et al. Haploinsufficiency of MSX1: a mechanism for selective tooth agenesis. Mol Cell Biol 1998;18:6044-51. [ Links ]
28. Wang H, Zhao S, Zhao W, Feng G, Jiang S, Liu W, et al. Congenital absence of permanent teeth in a six-generation Chinese kindred. Am J Med Genet 2000; 90:193-8. [ Links ]
29. Liu W, Wang H, Zhao S, Zhao W, Bai S, Zhao Y, et al. The novel gene locus for agenesis of permanent teeth (He-Zhao deficiency) maps to chromosome 10q11.2. J Dent Res 2001;80:1716-20. [ Links ]
30. Nieminen P, Arte S, Pirinen S, Peltonen L, Thesleff I. Gene defect in hypodontia: exclusion of MSX1 and MSX2 as candidate genes. Hum Genet 1995;96: 305-8. [ Links ]
31. Arte S, Nieminen P, Pirinen S, Thesleff I, Peltonen L. Gene defect in hypodontia: exclusion of EGF, EGFR, and FGF-3 as candidate genes. J Dent Res 1996;75:1346-52. [ Links ]
32. Ahmad W, Brancolini V, ul Faiyaz MF, Lam H, ul Haque S, Haider M, et al. A locus for autosomal recessive hypodontia with associated dental anomalies maps to chromosome 16q12.1. Am J Hum Genet 1998;62:987-91. [ Links ]
33. Slavkin HC. Entering the era of molecular dentistry. JADA 1999;130:413-7. [ Links ]
34. Gorlin RJ, Cohen MM Jr, Hennekam RCM, eds. Syndromes of the head and neck. 4th ed. New York: Oxford University Press; 2001. [ Links ]
35. Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 1996;13:409-16. [ Links ]
36. Pääkkönen K, Cambiaghi S, Novelli G, Ouzts LV, Penttinen M, Kere J, et al. The mutation spectrum of the EDA gene in X-linked anhidrotic ectodermal dysplasia. Hum Mutat 2001;17:349. [ Links ]
37. Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 1999;22:366-9. [ Links ]
38. Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001;27:277-85. [ Links ]
39. Tucker A, Headon D, Schneider P, Ferguson B, Overbeek P, Tschopp J, et al. Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development 2000;127:4691-700. [ Links ]
40. Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 1994;6:348-56. [ Links ]
41. Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, et al. Cloning and characterization of a novel bicoid-realated homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996;14:392-9. [ Links ]
42. Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T. et al. Identification of a Wnt/Dvl/beta-catenin-to-Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 2002;111:673-85. [ Links ]
43. Espinoza H, Cox C J, Semina EV, Amendt BA. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Molec Genet 2002;11:743-53. [ Links ]
44. Vieira AR. Oral clefts and syndromic forms of tooth agenesis as models for genetics of isolated tooth agenesis. J Dent Res 2003;82:162-5. [ Links ]
45. Sharpe PT. Homeobox genes and orofacial development. Connect Tissue Res 1995;32:17-25. [ Links ]
46. Gilbert S. Developmental biology. 6th ed. Massachussets: Sinauer Associates Inc. Sunderland; 2001. [ Links ]
47. Baum BJ, Mooney DJ. The impact of tissue engineering on dentistry. JADA 2000;131:309-18. [ Links ]
48. Line SRP. Molecular morphogenetic fields in the development of human dentition. J Theor Biol 2001;211:67-75. [ Links ]

