










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAvances en Periodoncia e Implantología Oral
versión On-line ISSN 2340-3209versión impresa ISSN 1699-6585
Avances en Periodoncia vol.18 no.2 Madrid ago. 2006
Interleukina 18, ¿nuevo factor a tener en cuenta en la patogénesis de la enfermedad periodontal?
Interleukin 18, A new factor in the pathogenesis of the periodontal disease
Andrés R.*, Amaro J.**, Bascones A.***
* Doctorando. Universidad Complutense de Madrid.
** Master en Periodoncia. Universidad Complutense de Madrid.
*** Catedrático de Medicina Oral y Periodoncia. Universidad Complutense de Madrid.
Dirección para correspondencia
RESUMEN
La enfermedad periodontal es un proceso infeccioso, crónico y de naturaleza inflamatoria, donde se produce la pérdida de los tejidos de sostén del diente.
Existe un acuerdo general en que son las bacterias de la placa las responsables del inicio y mantenimiento de la enfermedad; pero se cree que será el tipo de respuesta inmune del individuo, la responsable del grado de destrucción periodontal que se desarrolle. Se piensa que una respuesta mediada por anticuerpos produciría una mayor destrucción debido a la hiperestimulación del sistema inmune, mientras que una respuesta celular sería más protectora con los tejidos periodontales. En este artículo proponemos que, la interleukina 18 (IL-18), citokina inductora de interferón gamma, favorecería el desarrollo de una respuesta inmune celular, permitiendo una eliminación bacteriana más eficaz, reduciendo así el daño que sufrirían los tejidos periodontales.
Palabras clave: Interleukina 18, ICE, Th1, Th2, Interferón-
g, apoptosis.SUMMARY
Periodontal disease is an inflammatory, chronic and infectious process,where the tooth support tissues get lost.There is an agreement that bacteria are responsible for the beginning and support of the disease. It is believed that the type of individual immune response, is responsible for the periodontal destruction degree. It is thought that an immune response regulated by antibodies would produce a larger tissue destruction, due to the hyperstimulation of the immune system. While cellular immune response would be more protective with periodontal tissues.
In this paper,we suggest that interleukin 18 (IL-18), as an interferon gamma inductor,would favor the cellular immune response development, allowing a more efficient bacterial elimination, reducing the periodontal tissue damage.
Key words: Interleukin 18, ICE, Th1 y Th2, Interferon-g, apoptosis.
Introducción
La enfermedad periodontal se define como una enfermedad infecciosa crónica, cuya principal característica es la pérdida progresiva de los tejidos de sostén del diente. Esta pérdida puede darse de manera lenta y en brotes de destrucción espaciados en el tiempo, como sucede en la periodontitis crónica, o puede ser más rápida y severa como la periodontitis agresiva o de comienzo temprano.
Sus signos clínicos más importantes son el enrojecimiento del margen gingival, sangrado, pérdida de hueso alveolar, pérdida de inserción clínica, formación de bolsas periodontales y en casos avanzados movilidad dentaria y pérdida de los dientes.
Socransky y Haffajee en 1997 establecieron, mediante sus clusters bacterianos, que la infección es producida por bacterias del biofilm bacteriano, y que no todas ellas son capaces de inducir enfermedad (1). Se han conseguido aislar más de 500 especies bacterianas cultivables en el fluido crevicular de seres humanos (Moore & Moore 1994), pero podría haber más de mil (2), ya que hay muchas que no son clasificables o cultivables (Sockransky et al 1987).
A pesar de que bacterias periodontopatógenas ocasionan pérdida de inserción en determinados individuos, estas mismas bacterias no son capaces de inducir enfermedad en otros; esto es lo que llamamos susceptibilidad del individuo. La susceptibilidad viene determinada por una serie de parámetros como son la predisposición genética, virulencia bacteriana, enfermedades sistémicas o hábitos adquiridos como puede ser el tabaco (workshop 1996).
La microflora bacteriana periodontopatogénica es necesaria pero insuficiente para que se desarrolle la enfermedad, siendo necesario que el hospedador sea susceptible, y lo que marcará el carácter destructivo de la enfermedad será la naturaleza de la respuesta inflamatoria.
En la actualidad, cada vez se tiene mayor certeza que las bacterias no son responsables directas de la destrucción tisular como se pensaba antes. Estas pueden producir destrucción de los tejidos de dos maneras; de manera directa, invadiéndolos y produciendo diferentes toxinas capaces de inducir muerte celular y necrosis tisular; o de manera indirecta (cada vez cobra mayor importancia) como factor iniciador y posteriormente como responsable del mantenimiento de la respuesta inmune contra dicha infección. Las bacterias inducen una respuesta inflamatoria, y mientras persista esta colonización bacteriana, se mantendrá y aumentará la respuesta, siendo esta la que produzca el mayor grado de destrucción de los tejidos de sostén del diente.
La enfermedad periodontal tiene un marcado carácter autoinmune donde se produce la liberación de moléculas vasoactivas, enzimas proteolíticas, y diferentes sustancias por parte de las células de defensa que atacarán a las bacterias, pero que también dañaran los tejidos del individuo.
A pesar de las diferencias existentes en la etiología específica, progresión y momento de comienzo de todas las periodontitis, exhiben características clínicas e histopatológicas del proceso inflamatorio muy similares en todos los casos. Page & Schroeder en 1977 dividieron la patogénesis de la enfermedad periodontal en 4 etapas: inicial, temprana, establecida y avanzada.
Al existir una invasión bacteriana, las bacterias se sitúan adyacentes al epitelio de unión (EU), y será este la primera barrera física contra el progreso de la infección. Se produce una vasodilatación e inflamación de los vasos. Una de las primeras células inflamatorias que llegan a la lesión son los neutrófilos. Estos extravasan la pared de las vénulas postcapilares, migrando a través del EU, localizándose en el surco gingival, y anteponiéndose entre las bacterias y el EU. Según prolifera apicalmente la placa supragingival, las células del EU comienzan también a proliferar apicalmente a la superficie radicular, comenzando con así la formación de los primeros esbozos de la bolsa gingival.
En la formación del infiltrado, también participan los macrófagos, que van aumentando su número según el proceso evoluciona de gingivitis a periodontitis, y comienza a aumentar también el número de linfocitos T CD4+ Th1 y Th2. Coincide con el desarrollo y evolución de la enfermedad la aparición de células B, y según evoluciona, la bolsa periodontal va profundizando en dirección apical, destruyéndose los componentes de la matriz extracelular, ligamento periodontal y reabsorción de hueso alveolar.
Pero, en este complejo proceso de activación celular, ¿quién hace de mediador entre un grupo celular y otro?¿Quién decide si el tipo de respuesta que se va a desarrollar será tipo Th1 ó Th2.¿Qué o quién atrae a las células inmunitarias a la zona de la lesión?
La respuesta a estas preguntas es la misma. Son las interleukinas o citokinas.
Estas moléculas son polipéptidos o proteínas de bajo peso molecular, encargadas de iniciar y mantener la respuesta inflamatoria, regulando la amplitud y duración de esta. Son extremadamente potentes, y generalmente actúan a una concentración picomolar (3), interactuando con receptores presentes en la superficie de las células produciendo señales, y desencadenado diferentes acciones biológicas.
Probablemente las primeras células en actuar frente a las bacterias, podrían ser los epiteliocitos del EU, que producen IL-8, un potente factor quimiotáctico para los neutrófilos, que participarán de una manera significativa en la formación del infiltrado, siendo las primeras células en llegar a la lesión, y llegando incluso a formar una barrera física contra las bacterias. Una baja concentración de IL-8 estimulará la migración de estas células polimorfonucleares, mientras que una concentración mayor las activará.
Los macrófagos por otra parte tienen un papel central en la movilización de los mecanismos de defensa, participando en la presentación antigénica con funciones microbicida, tumoricida e inflamatoria. Expresarán en su superficie receptores que influencian la respuesta inmune antígeno específica que tendrá como célula diana las bacterias. Estos son estimulados por el lipopolisacárido de la membrana bacteriana, y que responderán secretando mediadores inflamatorios como IL-1b, PGE-2, y TNF-
a. Algunos de estos factores son importantes componentes implicados en la patogénesis de la enfermedad periodontal.A partir de aquí comienza un complejo proceso de activación de diferentes líneas celulares, con una actividad antibacteriana específica, mediante la presentación antigénica a los diferentes grupos de linfocitos; y estos, una vez activados y según el ambiente en el que se encuentren, comenzarán a liberar determinadas citokinas con diferentes funciones. Los linfocitos T CD4+ y los CD8+. Los primeros son también conocidos como linfocitos T helper o colaboradores; y los segundos como linfocitos citotóxicos.
A su vez los linfocitos T helper se dividen en Th1 y Th2, con un mismo precursor, que es el linfocito Th0 que se diferenciará en una de estas dos estirpes celulares.
Los Th1 se relacionan con actividad microbicida, activación y diferenciación de células T y macrófagos, mientras que los Th2 + favorecen la activación y diferenciación de células B.
La respuesta de una célula a una citokina, depende de la concentración local de esta, del tipo de célula que sea, u otros reguladores liberados en el medio.
Las interleukinas interactúan en una compleja red, induciéndose unas a otras, mediante sinergismos, antagonismos y diferentes funciones biológicas (3).
Por ejemplo la IL-1, probablemente la más estudiada, es una citokina pleiotrópica con una capacidad extremadamente potente para inducir reabsorción ósea mediante la activación de los osteoclastos, activar metaloproteinasas, produce neutrofilia, activa complemento, aumenta la expresión de moléculas de adhesión...
El interés de conocer las interleukinas no solo radica en el mero conocimiento de los procesos inmunológicos, sino en la capacidad a posteriori para controlar o regular diferentes enfermedades, mayormente autoinmunes, en las que estas pueden ser las responsables del inicio, mantenimiento y/o desarrollo.
Por ejemplo, la interleukina 18 se ha relacionado con diferentes enfermedades:
• Artritis reumatoide (AR) es una enfermedad asociada a un perfil de citokinas Th1. Se ha observado en estudios in vitro que macrófagos del sinovio producen IL-12. También se ha encontrado altas concentraciones de IL-18 en el fluido sinovial de pacientes con AR, producida también por los macrófagos y no por células T, aunque ambas expresan IL-18R (4). La IL-18, al ser un potente inductor de INF-g, podría mantener la respuesta inflamatoria de tipo Th1 (5).
• Diabetes mellitus tipo I, la inyección exógena de IL-18 inhibe el desarrollo de esta enfermedad en ratones diabéticos no-obesos (NOD). Sorprendentemente, el páncreas de ratón tratado con IL-18 expresó más IL-4 RNA que los no tratados; sugiriendo que las células T infiltradas en los islotes del páncreas cambian a células productoras de IL-4 (6). Esto puede ser una pista sobre la implicación de la IL-18 en las respuestas Th2.Varios autores han constatado que la IL-18 induce la producción de IL-4 e IL-13 por parte de células T, NK, mastocitos y basófilos (7). Recientemente se ha comprobado que la IL-13, que regula negativamente la producción de IL-18, es capaz de prevenir la diabetes en ratones.
• Esclerosis múltiple, donde se ha observado que, lesiones cerebrales de ratones expresan con mayor frecuencia y en mayor cantidad IL-18RNA e INF-gRNA que ratones sanos, y su infiltrado inflamatorio era preferentemente del tipo Th1. Además, sus monocitos periféricos expresaban también mayor concentración de caspasa-1 RNA. Se observó que los ratones que carecían de caspasa-1 (ICE) eran relativamente resistentes a este proceso autoinmune,ya que se reducía el desarrollo de esta respuesta Th1 (8, 9, 10).
Para colaborar en el conocimiento de este complejo sistema, vamos a centrarnos en la IL-18, que aunque no sido prácticamente estudiada a nivel periodontal, tiene numerosos estudios hechos a nivel de inmunología general, presenta características pleiotrópicas al igual que la IL-1.
Características
La IL-18 es una citokina pleiotrópica encargada de regular tanto la respuesta inmune innata, como la adquirida, y juega un papel fundamental en desórdenes autoinmunes, inflamatorios e infecciosos.
Fue descubierta en 1989, y fue denominada factor inductor de Interferón-g, ya que inducía la producción de este en células del hígado de ratones (11, 12).
Se clonó y caracterizo en 1995; y en 1996 fue relacionada y admitida como miembro de la familia de las IL- 1(13).
Semejanzas con la IL-1
IL-18 e IL-1 son proteínas estructuralmente homólogas, y comparten el mismo camino de transducción de la señal (14, 15).
A pesar de la importante similitud que presentan sus estructuras tridimensionales, solo comparten una homología del 15% en su cadena de amino-ácidos.
También poseen una misma enzima que las convierte en moléculas activas, que es la enzima ICE (también llamada caspasa-1), y ambas son citokinas pleiotrópicas.
Debido a las semejanzas y relaciones que presentan ambas interleukinas con los diferentes miembros de la familia IL-1, se ha sugerido que IL-1
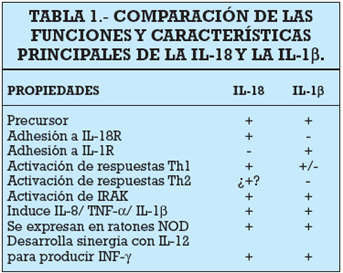
a, IL-1b e IL-18 podrían formar una sola subfamilia, hasta el punto que, en sus comienzos fue denominada IL-1g.Como se ha podido ver en diferentes estudios, las IL- 18 e IL-1b comparten propiedades y actividades biológicas (16), (tabla 1).
Células Productoras
Son muchas y diferentes las células productoras, o tejidos donde se localiza esta molécula; entre ellas se encuentran: macrófagos, células de Kupffer, células dendríticas, epiteliales, células microgliales (17, 18, 19, 20 ,21) y monocitos sanguíneos que la secretan como precursor inmaduro (pro-IL-18). Las células polimorfonucleares son capaces de secretar pro-IL-18 en condiciones normales, es decir, sin estímulo directo, (22) pudiendo ser activada extracelularmente.
Células osteoblásticas secretan IL-18, inhibiendo la diferenciación de las células osteoclásticas mediante la inducción de GM-CSF (Granulocyte Macrophage Colony Stimulating Factor). Se ha detectado también su secreción en nervios centrales (23) y sistema endocrino (24).
Estímulos que la inducen
La citokina es secretada ante ciertos estímulos, como por ejemplo la presencia o interacción de diferentes microorganismos o sus toxinas con las células productoras:
• Bacterias Extracelulares
La infección por ciertos patógenos extracelulares como puede ser el S. pyogenes, produce la liberación de citokinas como IL-18, IL-12 e INF-g por parte de las células polimorfonucleares (25).
La presencia de IL-18 se ha detectado en otras infecciones, como en el líquido cefalorraquídeo de pacientes con meningitis.
Ratones infectados por yersinia enterocolitica, y a los que previamente se ha neutralizado la actividad de su IL-18, desarrollan de 10 a 100 veces más cantidad de bacterias que los ratones control, sin encontrarse diferencias en la producción de interferón gamma, sugiriendo así el papel protector de la IL-18 por algún mecanismo independiente a la producción de Interferón gamma (26).
• Bacterias Intracelulares
Células epiteliales, desarrollan un perfil de citokinas Th1 en respuesta a una infección inducida por C. trachomatis. Esta, además, provocó la secreción inmediata de IL-18, sugierendo un papel importante en la defensa contra la bacteria. Los productos bacterianos son capaces de activar la enzima ICE y posteriormente producir la secreción de IL-18 activa (27).
Además una recombinación de IL-18 en total ausencia de INF-g favoreció la eliminación de la bacteria, poniendo de manifiesto el papel dominante de la IL- 18 sobre el INF-g (28).
En infecciones inducidas por S. typhimurium en ratones carentes de IL-18, a resistencia a ésta fue sensiblemente menor, y la carga bacteriana en bazo e hígado aumentó (29), demostrando así la importancia de IL-18 en la regulación de la respuesta inmune.
La IL-18 es requerida para la liberación de óxido nítrico por parte de los macrófagos en respuesta a microorganismos, y también demostró ser fundamental en respuestas memoria efectoras contra la reinfección bacteriana, acentuando así su papel en respuestas inmunes tanto primarias como secundarias.
• Virus
Se observó un papel protector de la IL-18 contra el virus herpes simple (HSV). La administración de IL-18 produjo interferón gamma,y este indujo liberación de óxido nítrico (NO), protegiendo al hospedador del virus, inhibiendo la replicación viral (29).
Macrófagos infectados por virus, produjeron IL-18, induciendo una respuesta Th1 por parte de los linfocitos T.
Varios tipos de virus, incluyendo la familia de los Poxvirus son capaces de producir proteínas homólogas a IL-18Bp, que se adhieren a la IL-18 inhibiendo su actividad biológica, principalmente la producción de interferón gamma (30, 31, 32).
Esto nos sugiere que el bloqueo de la IL-18 producido por las proteínas homologas a la IL-18Bp de los poxvirus podrían atenuar la respuesta inflamatoria antiviral.
Activación de la molécula
La activación de la molécula se produce intracelularmente; para ser biológicamente activa, debe ser procesada y pasar de una molécula de 24kDa a una de 18 kDa (33,34). El proceso de activación más estudiado es el relacionado con la enzima convertidora de IL-1; le enzima ICE (interleukin-1 converting enzyme) o Caspasa 1 (35), que divide la molécula entre los aminoácidos Asp35-Asn36, activando la molécula (36).
La trascendencia de la ICE se observó en estudios donde, ratones carentes de ICE y estimulados con LPS de P. acnes, no produjeron aumento de IL-18 ni INF-g, con consecuencias fatales.
El óxido nítrico (NO) puede suprimir la acción de la caspasa-1, resultando en una inhibición de secreción de IL-1-b e IL-18, siendo este un regulador negativo de ambas citokinas (37).Otras enzimas capaces de activar la IL 18 es la Caspasa-4 y la Caspasa-3, pero su eficiencia es de cien a mil veces más baja (38).
Se ha sugerido una posible activación extracelular de la molécula. Los monocitos sanguíneos secretan pro- IL-18 (39), y esta necesitaría ser activada extracelularmente, pero esto todavía no está muy claro; de hecho se ha encontrado el precursor libre en el medio extracelular; el cual podría provenir de la destrucción celular y posterior liberación al medio de la molécula.
Receptor y antagonismo de IL-18Bp
El IL-18R es un heterodímero compuesto por dos proteínas. La primera es el receptor de interleukina 18a (IL-18Ra), y tiene baja afinidad por la IL-18. La segunda es el receptor de interleukina 18b (IL-18Rb), también denominada AcPL (receptor de proteína accesoria) que no es capaz de unirse a la IL-18, pero aumenta sensiblemente la especificidad de dicha unión, y es esencial para la transducción de la señal (40,41,42).
El receptor de IL-18 se ha visto que solo es expresado en las células Th1 y no en las Th2, pudiendo ser un marcador de superficie para distinguir células Th1 de Th2 (43).
• IL-18Bp
La falta de un receptor antagonista de IL-18 podría ser compensada con la presencia de la IL-18Bp (Interleukin- 18 Binding protein) (44).
Esta molécula inhibe la producción de IFN-g mediante el bloqueo del receptor IL-18R por adhesión (45), actuando como una molécula antiinflamatoria e inmunosupresiva natural (46).
Funciones biológicas
Desempeña papeles importantes en el inicio y la regulación de la respuesta inmune (47) (tabla 2).
• Inducción de diferenciación de células Th1.
Se ha observado que sólo células T nativas con su TcR (T cell receptor) estimulado, y en presencia de IL-18, se desarrollaron a células Th1. La IL-18 por sí sola no es capaz de inducir esta diferenciación, pero sí acelera este proceso (48, 49).
• Induce la producción de Interferón Gamma
La IL-18, por sí sola es capaz de inducir la producción de pequeñas cantidades de INF-g in vivo (10) e in vitro al igual que la IL-12. Pero esta producción se dispara cuando actúan ambas citokinas de manera conjunta (9). Dicha inducción se ha observado en células NK, células dendríticas, microgliales, macrófagos, células T y B (50, 51).
• Aumenta la actividad citotóxica de los NK y de las células T CD8+
La IL-18 aumenta la citotoxicidad mediada por los NK. En este caso al estimularlas con IL-18 más IL-12, no se produce sinergismo (52, 53).
• Induce producción de IL-4
Células T CD4+ cultivadas in vitro con IL-18 e IL-2 producen modestas cantidades de IL-4; y por sí sola, la IL- 18 induce IL-13, y citokinas tipo Th2 (54).
• Papel protector de la IL-18 contra patógenos extra e intracelulares
La producción de INF-g inducida por la IL-18 protege al organismo de los microorganismos, y además fomenta una respuesta tipo th1.
• Papel regulador en la apoptosis
Se observó que la administración de IL-18 era capaz de activar las vías extrínseca e intrínseca de la apoptosis en diversas células, liberándose además otras citokinas proinflamatorias como TNF-
a e IL-1b (26).• Función antitumoral
Recientes estudios sugieren que la IL-18 tiene un papel importante en la acción anti-tumoral (54). Los macrófagos asociados a tumores han cobrado importancia debido a su capacidad de:
- liberar citokinas antitumorales como la IL-12a por inducir INF-g.
- aumentar de la citotoxicidad de las células CD8+ y las NK contra células tumorales.
• Otras funciones
- Regula la actividad de células Th1, de los Linfocitos T citotóxicos, los Natural Killers, macrófagos, células dendríticas y células B.
- Regula la producción de la enzima óxido nítrico sintetasa.
- Induce sobre células endoteliales la producción de moléculas de adhesión, como ICAM-1 y VCAM-1 (55).
- Posee capacidad para degradar cartílago.
- Aumenta la expresión de ciclo-oxigenasa 2 (COX-2).
- Induce la liberación de glucosaminglucanos.
- Activa las metaloproteinasas de la matriz (MMP´s).
- Inhibe la actividad de los osteoclastos mediante la inducción de factor estimulador de colonias de macrófagos/granulocitos (GM-CSF).
- Inhibe la angiogénesis (25).
- Posee propiedades quimiotácticas para atraer células polimorfonucleares mediante la inducción de IL- 8 (56, 57).
- Induce la producción de TNF-
a, y GM-CSF (58).- Colabora en los mecanismos de defensa, activando macrófagos y células T contra ataques microbianos (59, 60, 61, 62).
Discusión
Como hemos podido ver, la IL-18 es una citokina pleiotrópica, cuya función principal podría ser el inicio y mantenimiento de la respuesta inflamatoria. Según los estudios observados, esta molécula se comporta preferiblemente como un factor inductor de respuesta Th1; aunque dependiendo del ambiente en el que se encuentre, podría desarrollar contradictoriamente una respuesta Th2.
El hecho de que esta citokina desarrolle una respuesta tipo Th1, creemos que correspondería con el mantenimiento de una lesión periodontal protectora (gingivitis), en lugar de una respuesta más destructiva llevada a cabo por una respuesta tipo Th2. A lo largo de los últimos cuarenta años, ha habido disparidad de opiniones respecto a este tema, y a día de hoy, todavía no se ha logrado un consenso. Diferentes autores como Seymour y Greenspan 1979, Mackler et al 1977, protectora de la respuesta Th1. En dicho estudios se observa que el paso de gingivitis a periodontitis, se relaciona con un cambio en el infiltrado de la lesión; donde existía una respuesta dominada por linfocitos T, se pasa a una lesión más destructiva donde predominan células B/plasmocitos (68).
Por el contrario, autores como Ebersole y Taubman (1994), o Dennison y Van Dyke (1997), opinan exactamente lo contrario; una respuesta Th2, protegería al individuo de las bacterias periodontopatógenas (69- 71). Esta falta de consenso podría deberse a diversos factores, ya que la periodontitis es una enfermedad multifactorial.
Una respuesta Th2 en individuos susceptibles produciría una exagerada estimulación del sistema inmune, mediante el continuo marcaje de objetivos y células diana por anticuerpos ineficaces. Estos, hiperestimularían el sistema inmune sin ser capaces de marcar los objetivos correctamente, lo que produciría una gran liberación de enzimas que destruirán los tejidos de sostén del diente, favoreciendo la progresión de la lesión periodontal.
En lo que sí existe consenso, es en que son las citokinas las moléculas con un papel fundamental en determinar si la enfermedad periodontal progresa o se mantiene estable; el perfil de citokinas presentes en la lesión periodontal depende del tipo de linfocito presente.
La importancia de conocer los diferentes factores y ambientes que influyen en el comportamiento de esta citokina, radicaría en la posibilidad de poder manipularlos, y así poder modificar ciertas respuestas nocivas para el propio organismo. Observando los factores que actúan negativamente sobre esta citokina, podríamos conocer con más exactitud la susceptibilidad de un paciente a una determinada enfermedad; e incluso se podría manipular exógenamente dichos factores para anular o potenciar el desarrollo de determinadas respuestas.
La inducción de la IL-18 producir de INF-g supondría un mantenimiento de la respuesta Th1, a priori también facilitada por ella, manteniendo esta respuesta protectora para el individuo relacionada con una eliminación bacteriana más eficaz.
Bibliografía
1. Socransky SS, Haffajee AD. The nature of periodontal diseases. Ann Periodontol. 1997 Mar; 2(1):3-10. [ Links ]
2. Socransky SS, Haffajee AD, Smith GL, Dzink JL. Difficulties encountered in the search for the 3-3-3.
3. Kim SH, Eisentein M, Reznikov L, et al. Structural requeriments of six naturally occurring isoforms of the IL-18 Binding protein to inhibit IL-18. Proc Natl Acad Sci USA: 2000: 97: 1190-5.
4. Feldmann M, Brennan FM, Mini RN. Role of cytokines in rheumatois arthritis. Ann Rev Immunol 1996: 14: 397-440.
5. Dayer JM. Interleukin-18, rheumatoid arthtitis, and tissue destruction. J Clin Invest 1999; 104: 1337-9.
6. Rothe H, Haussmmann A, Casteels K, Okamura H, Kurimoto M, Bukart V, Mathieu C, Kolb H. IL-18 inhibits diabetes development in non-obese diabetic miceby counterregulation of Th1-dependent destructive insulitis. J immunol 1999: 163: 1230-6.
7. Hoshino T, Wiltrout RH, Young HA. IL-18 is a important coinducer of IL-13 in NK and T cells: a new potential role for IL-18 in modulating the immune response. J Immunol 1999: 1016-1041101-Saraneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A virus-induced IFN-b/
a and IL- 18 synergistically enhance INF-g gene expression in human T cells. J Immunol 1998: 160: 6032-8.8.Wildbaum G, Youseff S, Grabie N, Karin N. Neutralizing antibodies to INF-g-inducing factor prevent experimental autoimmune encephalomyelitis. J Immunol 1998: 161: 6368-74.
9. Leonard JP, Waldburger JE, Goldman SJ,. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med 1995: 181: 381-6.
10. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Hattori K et al. Cloning of a new cytokine that induces Interferon gamma production by T-cells. Nature 1995: 378: 88-91.
11. Imagen de interleukina 18. www.tsurumi.yokohamacu.ac.jp/.../pcenter.html
12. Imagen de interleukina-1b. www.cs.stedwards.edu/.../InterL/STRUC.HTML.
13. Bazán JF,Timans JC. A newly defined interleukin1? Nature 1996:379:591.
14. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S.Targeted disruption of the MyD88 gene results in loss of IL-1 and IL-18 mediated function. Immunity 1998: 9 143-50.
14. Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin 18 induces IL-8 and IL-1b via TNF-
a production from non CD-14+ human blood mononuclear cells. J Clinic Invest 1998: 101: 701-21.15. Poli G, Kinter AL, Justement JS, Bressler P, Kehrl JH. Retinoic acid mimics transforming grow factor beta in the regulation of human immunodeficiency virus exprtession in monocytic cells. Proc Natl Acad Sci USA 1992: 89: 2689-93.
16. Shapiro L, Puren AJ, Barton HA, Novick D, Peskind RL, Shenkar R, Gu Y, Su MS. Interleukin 18 stimulates HIV type 1 in monocytic cells. Proc Natl Acad Sci USA 1998: 22: 12550-5.
17. Cameron LA,Taha RA,Tsicopoulus A,Kurimoto M. Airway epithelium expresses IL-18. Eur respir J 1999:14 553-9.
18. Fantuzzi G, Puren AJ, Harding MW, Livingstone DJ. IL-18 regulation on interferon gamma production and cell proliferation as shown IL-1b- converting enzime(caspase-1)- deficient mice. Blood 1998: 91: 2118-25.
19. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A. Cloning of a new cytokine that induces Interferon gamma productions by T-cells. Nature 1995:378: 88-91.
20. Brieland JK, Jackson C, Hurts S, Loebenberg D, Muchamuel T, Debets R, Kastelkein R, Churakova T. Immunomodulatory role of endogenous interleukin-18 in gamma interferon mediated resolution of replicative Legionella pneumophila lung infection. Infect Immun 2000: 68: 6567-73.
21. Micallef MJ, Yoshida K, Kohno K, Ikeda M, Murimoto M. Interleukin 18 induces the sequential activation of Natural Killer cells and cytotoxic T lymphocytes to protect syngeneic mice for transplation with Meth A sarcoma. Cancer Research 1997: 57: 4557-63.
22. Micallef Mj,Yoshida K. Kawai S, HAnaya T, Kohno K, Arai S,Tanimoto T,Torigoe K. In vivo antitumor effects of murine INF g-inducing factor/IL-18 in mice bearing syngeneic Meth A sarcoma malignant ascites. Cabcer Immunol immunother 1997: 43: 361-7.
23. Prinz M, Hanisch UK.Murine microglial cells produce and respond to interleukin-18. J Neurochem 1999: 72: 2215-8.
24. Conti B, Jahng JW,Tinti C, Son JH, Joh TH. Induction of INF- g inducing factor in the adrenal cortex. J Biol Chem 1997: 272: 2035-7.
25. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Hattori K et al. Cloning of a new cytokine that induces Interferon gamma production by T-cells. Nature 1995: 378: 88-91.
26. Lu H, Shen C, Brunham RC. Chlamidya Trachomatis infection of epithelial cells induces the activation of caspase- 1 and release of of mature IL-18. J Immunol 2000:165: 1463-9.
27. Neighbors M, Xu X, Barrat FJ, Ruuls SR, Churakova T, Debets R, Bazan JF, Kastelein RA, Abrams JS O´Garra A. A critical role for interleukin 18 in primary and memory effectors responses to Listeria monocitogenes that extends beyond its effects on interferon gamma production. J Exp Med 2001: 194: 343-54.
28. Mastroeni P, Clare S, Khan S, Harrison JA, Hormaeche C, Okamura H, Kurimoto M, Dougan G. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulen Salmonella typhimurium. Infect Immun 1999: 67: 478-83.
29. Kim SH, Einstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, Dinarello CA. Structural requirements of six naturally occurring isoforms of the IL-18Bp to inhibit IL-18. Proc Natl Acad Sci USA 2000: 97: 1190-5.
30. EPuren AJ, Fantuzzi G, Dinarello CA,. Gene expresion, synthesis, and secretion of interleukin-18 and interleukin- 1b are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA 1999; 96; 2256-61.
31. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA. Interleukin 18 binding protein. A novel modulator of the Th1 cytokine response. Immunity 1999: 10: 127-36.
32. Xiang Y, Moss B. IL-18 binding and inhibition of interferon –gamma induction by human poxvirus-encoded proteins. Proc Natl Acas Sci USA: 1999:96: 11537-42.
33. Fantuzzi G, Reed DA, Dinarello CA. IL-12 induced INFgamma is dependent on caspase-1 processing of the IL- 18 precursor. J Clin Invest 1999: 104: 761-7.
34. Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G,Weidner J, Salley JP, Chaplin DD. Interleukin 1 converting enzyme requires aspartic acid residue for processing of the interleukin-1
b precursor at two distinct sites and does not cleave 31 kDa Il-1 alpha. J Immunol 1991: 147: 2964-9.35. Singer II, Scotts S, Chin J, Bayne EK, Limjuco G,Weidner J, Miller DK, Chapman K, Kostura MJ. The Interleukin-1- beta-converting enzyme (ICE) is localiced in the external cell surface membranes and in the cytoplasmatic ground substance of human monocytes by immuno-electron microscopy. J Exp Med 1995: 182: 1447-59.
36. Martin SJ, Green DR. Protease4 activation during the apoptosis; death by a thousand cuts? Cell 1995: 82: 349- 52.
37. Kim YM, Talanian RV, Li J, Billiar TR. Nitric oxid prevents IL-1b and INF-g-inducing factor (IL-18) release from macrophages by inhibiting caspase-1. J Immunol 1998: 161: 4122-8.
38. Fantuzzi G, Dinarello CA. The inflammatory response in interleukin-1b-deficient mice: comparison with other cytokine related knocked out mice. J Leukoc Biol 1996: 59: 489-93.
39. Li P, Allen H, Banerjee S, Seshadri T. Characterization of mice deficient in interleukin beta converting enzyme. J Cell Biochem 1997:64: 27-32.
40.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Flemming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, Kuriomoto M, Tanimoto T, Sato V, Flavell RA et al. Activation of Interferon-gamma inducing factor mediated by interleukin-1 beta converting enzyme. Science 1997: 275: 206-9.
41. Nicolas Delau, Mathias Bickel. Interleukin-1b and interleukin- 18: regulation and activity in local inflammation. Periodontology 2000: 2004: 35: 42-52.
42. Griffiths RJ, Stam EJ,Downs JT, Otternerss IG.ATP induces the release of IL-1 from LPS-primed cells in vivo. J immunol 1995: 154: 2821-8.
43. Parnet P, Garka KE, Bonnert TP, Dower SK, Sims JE. IL- 1Rrp is a novel receptor-like molecule similar to the type I interleukin-1 receptor and its homologues T1/ST2 and IL-1R AcP. J Biol Chem 1996: 271: 3967-70.
44. Prerregaus D, Gabel CA. Interleukin 1-b maturation and release in response to ATP and nigecerin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity . J Biol Chem 1994 : 269: 15195-203.
45.Yoshimoto T, Okamura H, Tagawa YI, Iwakura Y. Interleukin-18 together with Interleukin-12 inhibits IgE production by induction of Interferon gamma production from activated B cells. Proc. Natl Acad Sci USA 1997: 94: 3948-53.
46. Jiang X,Wang X. J Biol Chem 2000: 275: 31199-3120347- Matsumoto S, Tsuyi-Takayama K, Aizawa Y, Koide K. Biochem Biophis Res Commun 1997: 234: 454-7.
47. Akira S. The role of interleukin-18 in innate immunity. Current Opin Immunol 2000: 12: 59-6348-Bysani CH, Kirankumar V, Rama M, Min LW, Laurie B. Activation of intrinsic and extrisic proaptotic signaling pathways in interleukin-18 mediated human cardiac endothelial cell death. J Biol Chem 2004: 279: 20221-3.
48.Yoshimoto T, Takeda K, Tanaka T, et al. IL-12 upregulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for INF-g production. J Immunol 1998: 161: 3400-7.
49.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K. IL-12 upregulates IL-18 receptor expression on T cells, Th1 cells and B cells: synergism with Il-18 for INFg production. J Immunol 1997: 161: 3400-7.
50. Tominaga K,Yoshimoto T, Torigoe K, Kurimoto M, Matsui K. IL-12 synergises with Il-18 or IL-1b for Interferon Gamma production from human T cells. Int Immunol 2000: 12 (2) 151-60.
51. Boise L, González-García M, Postema CE, Ding L, Lindsten T, Turka LA. Cell 1993: 74: 597-608.
51b. Imagen de interferón gamma; www.biologictherapy. org/aboutbiotherapy/about.
52. Kohyama M, Saijyo K, Hayasida M, Yasugi T, Kurimoto M, Ohno T. Direct activation of human CD8+ cytotoxic T lymphocites by interleukin 18. Jpn J Cancer Res 1998: 89: 1016- 41.
53. Tomohiro Y, Hiroko T, Kouyi T, Katsuaki H, Haruki O, Shizuo A, William EP,KenjiN. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Allergy 1999: 96: 13962-6.
54. Born TL, Thomassen E, Bird TA, Sims JE. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J Biol Chem 1998: 273: 29445-50.
55. Chinnayan A, ORourkre, Tewari M, Dixit .FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis V. Cell 1995: 81: 505-12
55. Pan G, Risser P,Mao W, Baldwin DT, Zhong AW, Filvaroff E, Yansura D, Lewis L, Eigenbrot C et al. IL-1H, and interleukin- 1-related protein that binds IL-18R/ IL-1Rrp. Cytokine 2001: 13: 1-7.
56. Gracie JA, Robertson SE, McInes IB. J Leukocite Biol 2003. 73: 213-24.
57. O´Neil LA, Dinarello CA. The IL-1 receptor/toll like receptor superfamily: crucial receptors for inflamation and host defense. Immunol Today 2000: 21: 206-9.
58. O´neill LA, Greene C. Signal transduction pathways activated by the IL-1 receptor family: ancient signalling machinery in mammals, insects and plants. J leucoc Biol 1998: 63: 650-7.
59. Fujioka N,Akazawa R,Ohashi K,Fuiji M,Ikeda M,Kurkimoto M. Interleukin 18 protect mice against acute herpes simplex virus type 1 infection. J virol 1999: 73: 2401-9.
60. Maestroni P, Clare S, Khan S, Harrison JA, Hormaeche CE et al. IL-18 contributes to host resistance and gammainterferon production in mice infected with Salmonella Tiphimurium. Infect immune 1999: 67: 478-83.
61. Okamoto I, Kohno K, Tanimoto T, Ikegami H,Kurimoto M. Development of Cd8+ effector T cells is differentially regulated by IL-18 and IL-12. J Immunol 1999162: 3202-11
61. Akira S. The role of IL-18 in innate immunity. Cuur Opin Immunol 2000: 12: 59-63.
62. Adachi O, Kawai T,Takeda K, et al.Targeted disruption of the MyD88 gene results in loss of IL-1 and IL-18-mediated fuction. Immunity 1998; 9: 143-50.
63. Mackler BF, Frostad KB, Robertson PB, Levy BM (1977). Immunoglobulin bearing lymphocytes and plasma cells in human periodontal disease. J Periodontal Res 12:37-45
64. Seymour GJ,Greenspan JS (1979).The phenotypic characterization of lymphocyte subpopulations in established human periodontal disease. J Periodontal Res 14:39-46.
65. Lindhe J, Liljenberg B, Listgarten M (1980). Some microbiological and histopathological features of periodontal disease in man. J Periodontol 51:264-9.
66. Seymour GJ, Powell RN, Aitken JF (1983). Experimental gingivitis in humans. A clinical and histologic investigation. J Periodontol 54:522-8.
67. Reinhardt RA, Bolton RW, McDonald TL, DuBois LM, Kaldahl WB (1988). In situ lymphocyte subpopulations from active versus stable periodontal sites. J Periodontol 59:656-670. 204-Reinhardt RA,
68. Bolton RW, McDonald TL, DuBois LM, Kaldahl WB (1988). In situ lymphocyte subpopulations from active versus stable periodontal sites. J Periodo205- Taubman MA, Haffajee AD, Socransky SS, Smith.
69. DJ, Ebersole JL (1992). Longitudinal monitoring of humoral antibody in subjects with destructive periodontal diseases. J Periodontal Res 27:511-21.
70. Ebersole JL, Cappelli D, Sandoval MN, Steffen MJ (1995). Antigen specificity of serum antibody in A. actinomycetemcomitans- infected periodontitis patients. J Dent Res 74:658-66.
71. Ebersole JL, Taubman MA (1994). The protective nature of host responses in periodontal diseases. Periodontol 2000 5:112-41.
![]() Dirección para correspondencia
Dirección para correspondencia
Rodrigo Andrés García.
C/Capitán Haya, 51-19º B. 28020 Madrid
Tel: 670700997
e-mail: randgar@gmail.com
Aceptado para publicación: Abril 2005

