










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La hipertensión arterial pulmonar (HAP) es una patología de baja prevalencia (1,5/100.000 habitantes); por lo que está clasificada como enfermedad rara. HAP se define como grupo de enfermedades caracterizadas por un aumento progresivo de la resistencia vascular pulmonar (RVP) dando lugar a insuficiencia ventricular derecha y muerte precoz1. Cursa con aumento de presión arterial pulmonar media (PAPm) ≥25 mmHg medido por cateterismo cardiaco derecho (CCD)2. Tras el congreso de Niza 2018 se modifica el punto de corte de la PAPm >20 mmHg y la resistencia vascular pulmonar ≥3 Unidades Wood. HAP se clasifica en cinco grupos según la “European Society of Cardiology/European Respiratory Society 2015”3. Debido a su baja prevalencia, carácter crónico y progresivo, está asociada a mal pronóstico y alta mortalidad. Las recomendaciones de tratamiento en monoterapia se basan en bloqueantes de canales de calcio, antagonistas de receptores de endotelina (ARE), inhibidores de la fosfodiesterasa 5 (IPDE5), estimuladores de la guanilato ciclasa soluble (GC), análogos de prostaciclina y agonistas del receptor de prostaciclina (receptor IP). Desde la introducción de los últimos tratamientos ha mejorado la esperanza de vida, pero el pronóstico a largo plazo sigue siendo malo con una elevada mortalidad (40% a los tres años)4.
Por todo ello, hemos estudiado parámetros clínicos como la variación en el test de la marcha de 6 minutos (PM6M), PAPm, el porcentaje de saturación de oxígeno y capacidad funcional (CF) según los fármacos. También hemos revisado la seguridad de los fármacos específicos para su tratamiento y la adherencia terapéutica.
METODOLOGÍA
Estudio descriptivo observacional trasversal y retrospectivo realizado en un hospital de tercer nivel entre mayo de 2004 y agosto de 2020.
Se incluyeron pacientes mayores de 18 años con diagnóstico de HAP clasificados por la European Society of Cardiology/European Respiratory Society3, que acudieron a las consultas de farmacia a recoger el tratamiento para HAP. Los fármacos dispensados son: sildenafilo, tadalafilo, bosentán, ambrisentán, macitentán, riociguat, selexipag, treprostinil.
Se excluyeron pacientes sin datos clínicos recogidos en la historia electrónica clínica.
Para la recogida de datos demográficos y clínicos se emplearon la historia clínica y el programa de prescripción electrónica.
Las variables demográficas recogidas fueron: edad, sexo y fecha de nacimiento. También se recogieron datos clínicos como diagnóstico, edad al diagnóstico, enfermedades concomitantes, servicio responsable, fecha última revisión y tiempo entre las dos últimas consultas.
Las variables clínicas para el diagnóstico y valoración de HAP recogidas fueron: CF, porción N-terminal del péptido natriurético cerebral (pro-BNP), PAPm, PM6M, capacidad de difusión pulmonar (DLCO), medida de excursión sistólica del anillo tricúspide (TAPSE), CCD y el riesgo según la CF. Se recogieron estas variables en el momento del diagnóstico y en la última revisión.
Además, se recogieron los datos respectivos al tratamiento: principio activo, dosis, posología, fecha de inicio y fecha fin, número de fármacos prescritos (mono/bi/triterapia), grado de recomendación según nivel de evidencia y efectos adversos. Además se midió la adherencia a través de la tasa de posesión de medicación (TPM).
El análisis estadístico se realizó mediante el programa IBM-SPSS Stadistics®. Se consideró un nivel de confianza del 95%, aceptándose como significativas las diferencias encontradas con valor p<0,05. Para la descripción de las variables cuantitativas se utilizó la media y desviación estándar para variables con distribución normal y mediana y amplitud intercuartil para variables con distribución no normal. Para la descripción de variables cualitativas, se calcularon las frecuencias y porcentajes. Para relacionar variables cualitativas se utilizó Chi-cuadrado y para comparar el rango medio de las variables cualitativas con cuantitativas apareadas y siguiendo una distribución no normal se utilizó Wilcoxon.
El estudio fue aprobado por el Comité Ético de Investigacion con Medicamentos de La Rioja (CEImLAR) en diciembre de 2020.
RESULTADOS
Se incluyeron 27 pacientes (22 mujeres). La media de edad fue de 62,6±16,3 años y al diagnóstico 54,4±17,3 años. Ningún paciente falleció durante el estudio, siendo la media de tiempo de evolución de la enfermedad desde el diagnóstico de 96,4±58,2 meses. En el grupo de HAP idiopática (HAPi) fue 107,6±41,7 meses y el grupo HAP debido a enfermedad del tejido conectivo fue 85±54,7 meses.
Dos pacientes (7,4%) se clasificaron como grupo 1.1 (HAPi), 12 (44,4%) grupo 1.4.1 (enfermedad del tejido conectivo asociado a HAP); tres (11,1%) grupo 1.4.4 (enfermedad cardiaca congénita); dos (7,4%) grupo 3 (hipertensión pulmonar por enfermedades pulmonares y/o hipoxemia); cuatro (14,8%) grupo 4 (hipertensión pulmonar tromboembólica crónica: HPTEC) y cuatro (14,8%) grupo 5 (HAP con mecanismos poco claros o multifactoriales).
Del total de pacientes, 22 (81,5%) presentaban enfermedades concomitantes. El 37% presentaban una única enfermedad concomitante, 14,8% presentaban dos y 29,6% tres o más. Estas se clasificaron en función del sistema al que afectan. Dentro del sistema circulatorio y cardiaco ocho pacientes (29,6%) presentaban hipertensión arterial, cuatro (15,9%) fibrilación auricular, tres (11,1%) insuficiencia cardiaca isquémica, tres (11,1%) cardiopatía congénita, uno (3,7%) trombosis y otro (3,7%) anemia. Los pacientes que padecían enfermedades del tejido conectivo fueron tres (11,1%), además dos (7,4%) presentaban lupus eritematoso sistémico, otros dos (7,4%) síndrome de Crest y uno (3,7%) amiloidosis cutánea. Dentro de las que afectan al sistema respiratorio, dos pacientes (7,4%) presentaban enfermedad obstructiva crónica (EPOC), uno (3,7%) asma y otro (3,7%) síndrome de apneahipopnea durante el sueño. Por último, nueve (33,3%) presentaban comorbilidades asociadas al sistema endocrino: cinco (18,5%) diabetes mellitus tipo 2; cuatro (14,8%) dislipemia y obesidad y uno (3,7%) hipotiroidismo.
El servicio al que correspondían el mayor porcentaje de pacientes fue neumología (74,1%), luego cardiología (11,1%), medicina interna (7,4%) y reumatología (7,4%). La media del periodo entre consultas fue de 4,8±2,4 meses. El tiempo entre el diagnóstico y la última valoración varía entre 1 y 14 años. Los datos se recogen en la tabla 1.
Al comparar los valores clínicos de inicio y del último registro no se obtuvieron diferencias estadísticamente significativas por el test de Wilcoxon (PM6M, CF, proBNP, PAPm, porcentaje de mejora en PM6M, el porcentaje de saturación de oxígeno en sangre en función del grupo de fármaco, sexo o grupo de edad de los pacientes).
Tabla 1. Parámetros recogidos al diagnóstico y final del estudio de los informes clínicos para valorar la hipertensión arterial pulmonar

N*: número de pacientes; **: escala de disnea modificada Borg7.
Al comparar la distancia recorrida en la última PM6M recogida y en la primera, se obtienen diferencias en función del sexo (p=0,028). No se obtuvieron diferencias por el test de Chi-cuadrado en función del grupo de edad, grupo de fármacos y diagnóstico. La variación de los parámetros medidos en el primer y último registro se expresa en la tabla 2.
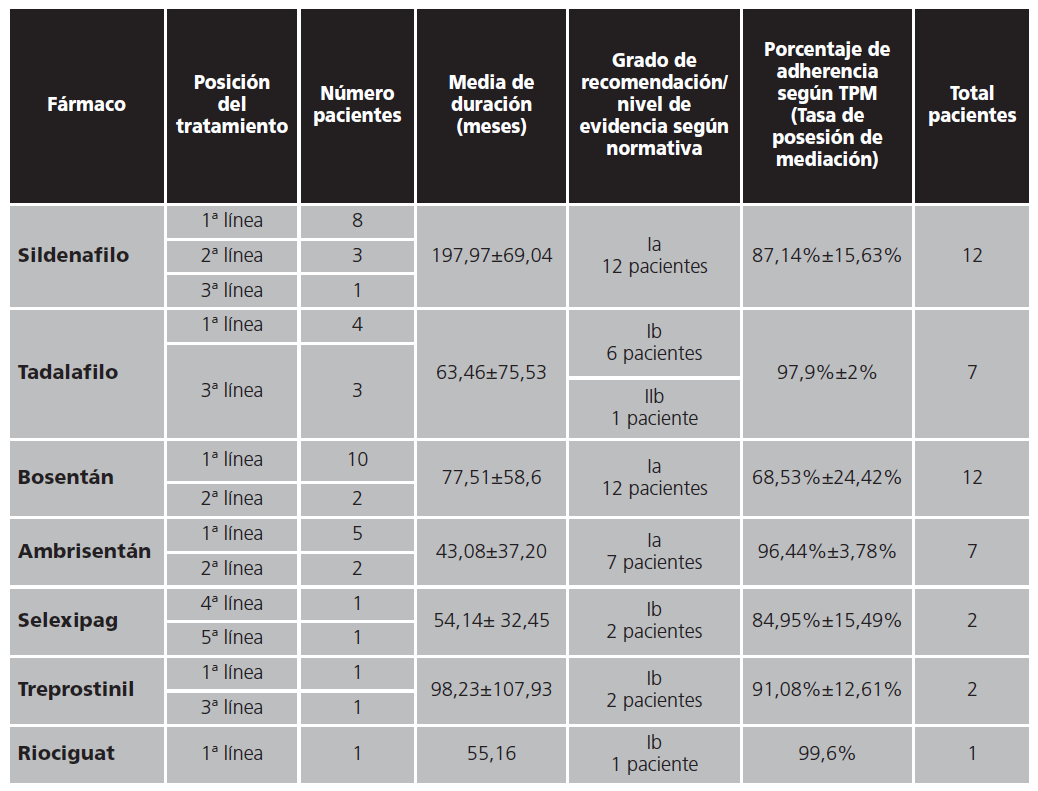
El tratamiento específico fue monoterapia en 21 pacientes (77,8%), biterapia en dos (7,4%) y triterapia en cuatro (14,8%). Los datos de cada fármaco en monoterapia se presentan en la tabla 3.
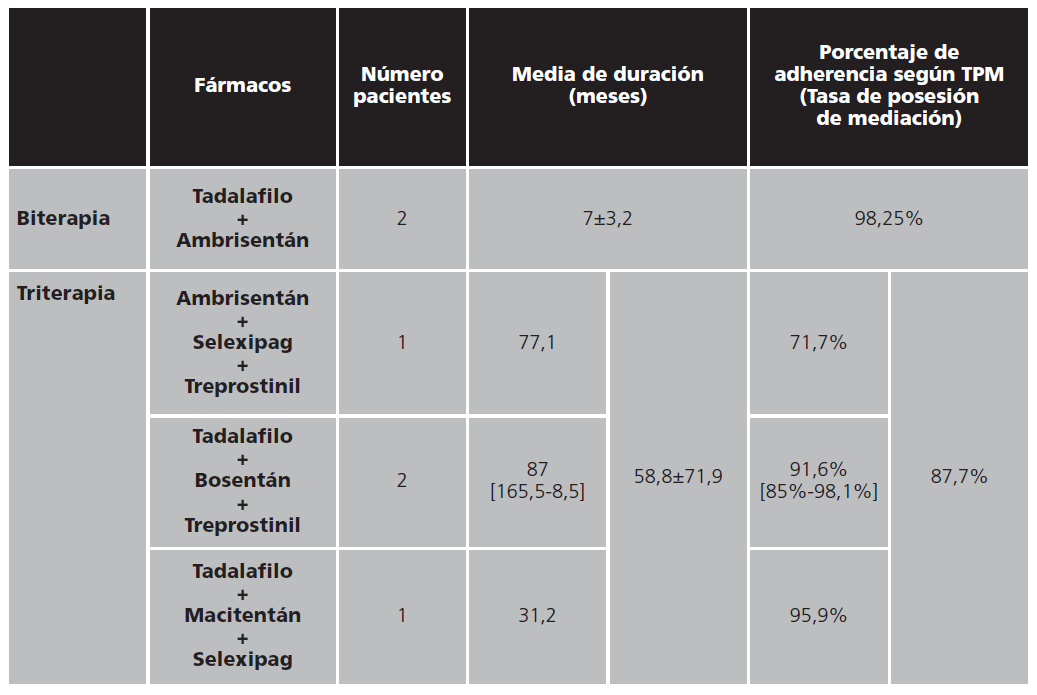
Las características de la bi y triterapia se muestran en la tabla 4.
Las indicaciones de la ficha técnica (FT) se cumplieron en el 88,9% de los pacientes. En cuanto a la seguridad de estos fármacos, sólo cuatro pacientes (14,8%) sufrieron algún efecto adverso. Dos (7,4%) presentaron rubefacción y cefalea con tadalafilo, uno (3,7%) rubefacción, cefalea, obstrucción nasal y dolor en extremidad inferior con selexipag y otro (3,7%) náuseas con selexipag. La gravedad de los efectos adversos fue leve, y se resolvieron al reducir la dosis. Ninguno de los pacientes tuvo que suspender el tratamiento.
DISCUSIÓN
El grupo de HAP mayoritario en la población española es el grupo 1, siendo el subtipo HAPi el 30% y HAP asociada a conectivopatías el más frecuente de HAP asociado a una enfermedad5. La prevalencia de HAP en el grupo 3 aumenta con la fibrosis pulmonar y el enfisema siendo mayor a un 50% en EPOC avanzado6,7. Según el registro español de hipertensión arterial pulmonar (REHAP) el grupo 4 supone un 15% de los pacientes8.
En el REHAP es mayoritario el grupo de cardiopatía congénita seguido de enfermedades del tejido conectivo (17,5% y 16% respectivamente) y un 12,3% otras causas como VIH e hipertensión portal. En un estudio realizado en 2007 el subtipo más frecuente fue grupo 2 (78,7% de los pacientes con cardiopatía izquierda), seguido de grupo 3 (enfermedades pulmonares 9,7%), grupo 1 sólo un 4,2% y grupo 4 un 0,6% (14). Sin embargo, en un estudio del Miguel Servet el tipo mayoritario fue la HAPi (37,1%), seguido del HTEPC (31,4%) y la HAP asociada a conectivopatía un 22,9%4.
Comparándolo con nuestro estudio, el subgrupo mayoritario fue el asociado al tejido conectivo (44,4%), muy por encima de la prevalencia de los estudios anteriores. Esto es debido a que los pacientes con cardiopatías se derivan a otros hospitales de referencia. Los segundos tipos más frecuentes fueron HPTEC y asociada a mecanismos desconocidos (14,8% cada una), seguida de HAP asociada a cardiopatía congénita (11,1%). HAPi y HAP secundaria a enfermedad pulmonar el 7,4%, muy por debajo del porcentaje obtenido en los anteriores estudios.
Tabla 2. Variación de los parámetros clínicos registrados al diagnóstico y el final del estudio
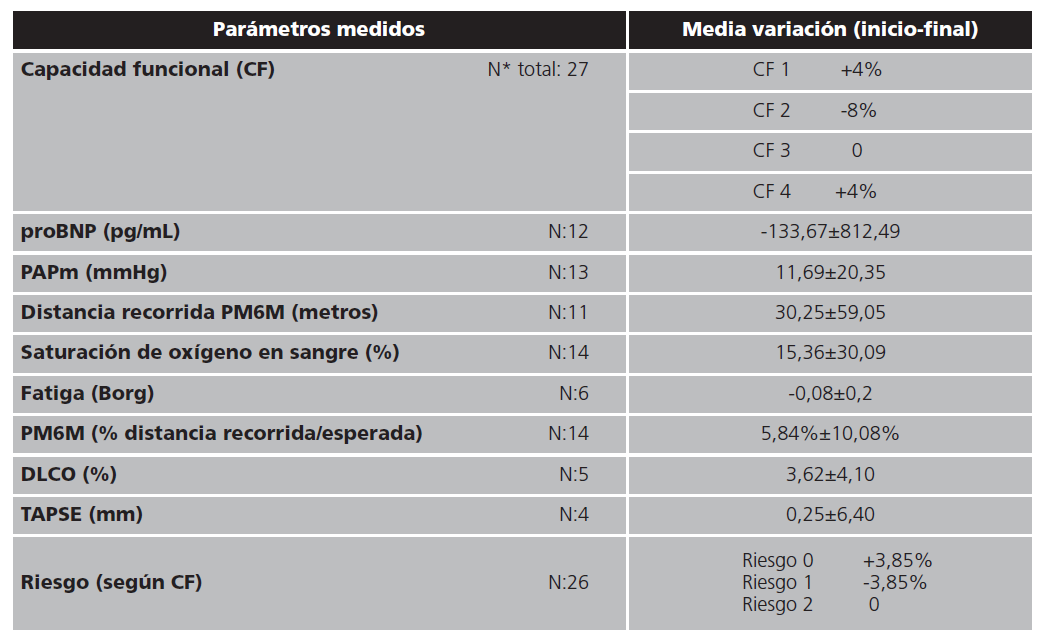
N*: número de pacientes.
Tabla 3. Tratamiento farmacológico específico en monoterapia de los pacientes de hipertensión arterial pulmonar
La población de nuestro estudio presentaba enfermedades concomitantes, muchas asociadas a HAP como conectivopatías (ocho pacientes), cardiopatías (seis pacientes) y enfermedades respiratorias (cuatro pacientes). Según el registro REVEAL9 más de la mitad de los pacientes presentaban dos o más comorbilidades, siendo mayoritaria la hipertensión arterial (32%) al igual que en nuestro estudio (29%). El hipotiroidismo y la esclerodermia representaban el 15%; similar en cuanto a esclerodermia en nuestros pacientes (11%). Estas patologías están estrechamente relacionadas con HAP con el aumento de la presión por la mayor resistencia de los capilares pulmonares.
El diagnóstico de la HAP es fundamentalmente clínico, se efectúa en edades avanzadas y se encuentra subdiagnosticada10, no es el caso de nuestros pacientes que están diagnosticados con grupo y categoría de HAP. En nuestro estudio el servicio mayoritario es neumología, pero hay que destacar que al no ser un hospital de referencia de HAP no realizan todas las pruebas diagnósticas, aunque sí realizan seguimiento regular y control del tratamiento. En el consenso11 de la Sociedad de Neumología y Cirugía Torácica y de Cardiología, se establece un tiempo mínimo de consultas de 6 meses, se deben repetir la PM6M, analítica sanguínea, electrocardiograma, radiografía de tórax y examen físico12. Estas pruebas se realizan en la unidad de referencia, pero es necesario que haya una buena coordinación entre esta y el hospital del paciente debido a la rápida progresión de la enfermedad. En nuestro estudio el tiempo de consultas es menor a 5 meses por lo que se encuentra dentro de las recomendaciones, pero los informes del hospital de referencia no siempre están disponibles, ya que los sistemas informáticos no son interoperables.
El sexo femenino es un conocido factor de riesgo para HAP, en los registros nacionales la proporción de mujeres: hombres es próxima a 2 debido a que en HAP hereditaria el riesgo de mutación del gen es un 42% en mujeres y un 14% en hombres13. En nuestro estudio, el ratio mujeres: hombres (4,4:1) es muy superior. El 100% de los pacientes hombres de nuestro estudio recorren más metros en el último PM6M pero solo un 20% de las mujeres mejoran, lo cual alcanzó significación estadística.
En España, la edad media de aparición es próxima a los 50 años11, similar a nuestro estudio, 55 años.
En cuanto a los factores pronósticos no existe evidencia científica suficiente para establecer valores óptimos para los parámetros que definen la evolución de los pacientes con HAP14.Se conoce que el pronóstico de HAP asociada a esclerosis es 2,9 veces peor que HAPi9,similar a nuestro estudio. Los meses de evolución de enfermedad son mayores en HAPi que en HAP asociada a esclerosis, por lo que se estima mayor duración de la enfermedad, pero no que tengan mayor supervivencia, ya que no se ha medido. Uno de los parámetros pronósticos utilizados es la CF de la OMS: clases I, II se relacionan con buen pronóstico y IV mal pronóstico3,4,15,16. En un registro francés17, el 75% de los pacientes fueron diagnosticados con CF IIIIV. En nuestro estudio más del 80% de los pacientes fueron diagnosticados con CF II, III, menos del 10% CF IV. Al tratarse de una enfermedad progresiva cabe esperar que la CF empeore con el paso del tiempo y de los síntomas. En el caso de nuestros pacientes no presentaron mucha variación: un paciente disminuyó CF de IV a III y otro empeoró de CF I a II.
Tabla 4. Tratamiento farmacológico específico en combinación (biterapia o triterapia) de los pacientes del estudio para hipertensión arterial pulmonar
También es un factor pronóstico la PM6M: mayor a 500 m buen pronóstico y menor a 300 m mal pronóstico3,4,15,16. En el caso de nuestros pacientes la media se sitúa entre estos dos valores adecuándose a un riesgo intermedio. El TAPSE también se relaciona con el pronóstico, siendo la media de nuestra población muy similar entre el diagnóstico y el último registro, aunque menor de 2 cm por lo que se sitúa en el límite del mal pronóstico (<1,5 cm). Las concentraciones plasmáticas de BNP/NT-proBNP también se asocian a peor pronóstico si son mayores a 1.400 ng/L. La población de nuestro estudio presenta valores intermedios. Por último la PAPm se utiliza como punto de corte para definir HAP, luego todos presentan PAPm >20 mmHg.
El tratamiento para HAP se basa en recomendaciones generales y de soporte3. Los pacientes de nuestro estudio estaban en fases más avanzadas o presentaron prueba vasodilatadora negativa por lo que llevaban tratamiento específico. Las recomendaciones establecen que las CF II, III comiencen con monoterapia o combinación si no hay respuesta y los pacientes con CF IV utilicen epoprostenol en monoterapia o en combinación3.
Los fármacos más utilizados en nuestro estudio fueron sildenafilo y bosentán en 12 pacientes. La mayoría en primera línea, pero también en líneas posteriores cuando no hubo respuesta clínica con otros fármacos. Ambos fármacos están indicados para CF II, III, con grado de recomendación Ia18,19.
En segundo lugar se han utilizado el tadalafilo y ambrisentán. Esto resulta lógico ya que los primeros son los fármacos que más evidencia y estudios tienen20, y al no existir estudios de comparación directa entre los fármacos del mismo grupo21 se recomienda priorizar a sildenafilo y bosentán en cada grupo.
Por último, los fármacos minoritarios en nuestra población fueron selexipag, treprostinil y riociguat. Son los fármacos de más reciente comercialización con indicación para HAPi y hereditaria, asociada a tejido conjuntivo y cardiopatía congénita con grado de recomendación Ib, como los pacientes de nuestro estudio. El único con indicación para HPTEC22 es el riociguat con grado de recomendación según la CF de Ib para CF II-III como es el caso del paciente de nuestro estudio.
Los fármacos en combinación utilizados por dos pacientes (7,4%) fueron tadalafilo y ambrisentán única combinación que ha demostrado superioridad al tratamiento en monoterapia23 con una media de 7±3,12 meses de tratamiento. La media de duración con estos fármacos en combinación es menor a los fármacos en monoterapia, ya que a medida que los pacientes empeoran se pasa de monoterapia a bi o triterapia.
La importancia del tratamiento específico para estos pacientes radica en la disminución de la mortalidad y mejora de la sintomatología sobre todo la disnea. La eficacia en los ensayos clínicos se midió con PM6M. Al relacionar la variación de PM6M con los distintos fármacos no se obtuvieron diferencias significativas, posiblemente por el bajo tamaño muestral y la falta de datos completos de los pacientes. La variable secundaria utilizada fue PAPm. En nuestro estudio la variación de PAPm es positiva ya que la media al diagnóstico es mayor que la media del último registro. En el caso de sildenafilo la disminución de la PAPm fue de hasta 5,1 mmHg para una dosis de 80 mg al día18 y bosentán disminuyó 8 mmHg19, frente a la disminución de 11 mmHg en el total de nuestros pacientes. Los valores de proBNP, saturación de oxígeno, fatiga, DLCO, metros esperados y TAPSE no presentan mejoría comparando las dos mediciones.
Por último cabe destacar que no ha habido ningún fallecido durante el estudio. Además los 27 pacientes llevan más de tres años con tratamiento específico de HAP, por lo que el pronóstico es mejor que el mostrado en estudios anteriores ya mencionados.
En la mayoría de los pacientes, el diagnóstico se corresponde con las indicaciones aprobadas. Hay un pequeño porcentaje que no cumple estas indicaciones. Cuando la monoterapia no es efectiva se asocian los fármacos del mismo o distinto grupo o se cambia de grupo, siendo el caso de los pacientes con HAP asociada a tejido conectivo que solo tiene indicación el ambrisentán, y nuestros pacientes llevan otros tratamientos.
Es importante medir la adherencia de estos pacientes ya que en muchos casos la posología y el número de medicamentos complican la toma correcta de los mismos. Los pacientes de nuestro estudio muestran adherencias elevadas, siendo la más baja alrededor del 69% con bosentán. El único método utilizado para medir las adherencias es la TPM, sería necesario realizar cuestionarios de adherencia para completar el estudio. Además dado que son pacientes con una enfermedad crónica es necesario reforzar la adherencia, mantener la motivación del paciente y el compromiso del paciente con su tratamiento por su beneficio en cuanto a supervivencia y mejoría de síntomas.
Todos los efectos adversos presentados estaban descritos en FT. En el caso de tadalafilo la rubefacción y cefalea está descrita como efecto adverso frecuente23 y con selexipag se encontraban descritos como muy frecuentes tanto la cefalea, rubefacción y dolor de extremidades24. Al ser de grado leve se resolvieron con la disminución de dosis y no tuvieron que suspender el tratamiento.
CONCLUSIONES
- Las variables de efectividad no muestran mejoría significativa estadísticamente, debido al bajo tamaño muestral. Si se tiene en cuenta el periodo de tratamiento de los pacientes, estos presentan esperanzas de vida mayores a tres años desde el inicio del tratamiento específico.
- Los efectos adversos encontrados han sido escasos y leves, por lo que se puede concluir que son fármacos seguros que se pueden manejar por los servicios encargados de estos pacientes sin tener que suspender el fármaco.
- La adherencia de los pacientes a su tratamiento es elevada, aunque se deben realizar cuestionarios específicos, validados y de forma frecuente.
LIMITACIONES
El estudio presenta una serie de limitaciones que hay que tener en cuenta como el pequeño tamaño muestral. El sesgo de infraregistro, ya que la historia clínica no está diseñada para recoger los datos específicos de esta patología. Además, al no ser un centro de referencia de tratamiento de HAP no constan todos los datos e informes necesarios.

