










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSanidad Militar
versión impresa ISSN 1887-8571
Sanid. Mil. vol.70 no.2 Madrid abr./jun. 2014
https://dx.doi.org/10.4321/S1887-85712014000200004
ARTÍCULO ORIGINAL
Validación in-house y determinación de grasa por gravimetría en conservas de raciones de combate del Ejército de Tierra
Validation in-house and uncertainty estimation by recovery assays in fat gravimetric determination in canned foods of combat rations Spanish Army
Pérez Grana R.
Tcol. Veterinario. Centro Militar de Veterinaria de la Defensa. Servicio de Bromatología y Seguridad Alimentaria. Madrid. España.
Dirección para correspondencia
RESUMEN
Objetivos: Validación in-house y estimación de la incertidumbre mediante ensayos de recuperación en la determinación de grasa en conservas de raciones de combate, utilizando la información generada en el proceso de validación.
Material y métodos: La determinación de la grasa se realiza por gravimetría, previa hidrólisis con ácido clorhídrico y extracción de la grasa con éter de petróleo 40-60o, mediante el sistema Soxtec System Tecator. Se analizan matrices y concentraciones (baja, media y alta) representativas de las muestras de rutina utilizando un diseño anidado de tres factores. La veracidad del método se evalúa estimando el sesgo proporcional a partir de los ensayos de recuperación. La estimación de la incertidumbre se realiza aplicando la ley de propagación de la incertidumbre, agrupando términos relativos de incertidumbre de precisión, recuperación y pureza del estándar. La incertidumbre expandida se obtiene aplicando un factor de cobertura K=2 (nivel de confianza del 95%).
Resultados: Se obtienen recuperaciones medias de grasa entre 0,95 y el 1,01 en tres niveles de concentración, por lo que la exactitud del método se considera buena. Los porcentajes de incertidumbre expandida relativa que se declaran para cada nivel de concentración de grasa son los siguientes: 0,50% (17,64%); 0,5-1,00% (13,09%); 1,00-16,55% (6,62%); 16,55-35,14% (6,00%); 35,14-50% (7,00%).
Conclusiones: La incertidumbre se estima utilizando la información generada durante el proceso de estimación del sesgo proporcional y puede aplicarse al análisis de futuras muestras de rutina con este método analítico.
Palabras clave: Incertidumbre. Recuperación. Grasa. Conservas. Raciones de combate. Ejército de Tierra.
SUMMARY
Objectives: Validation in-house and estimation of the uncertainty through recovery assays in determination of fat in canned foods of combat rations by gravimetríc using the information generated in the validation process.
Material and methods: Fat determination is carried out by gravimetríc, previously hydrolyzed with hydrochloric acid and extraction of fat with ether of petroleum 40-60o, by means of Soxtec System Tecator. Representative matrix and concentrations of the future routine samples are analyzed (low, medium, high) by using a nested design of three factors. The trueness of the method is evaluated by estimating the proportional bias starting from the recovery assays. The uncertainty estimate is carried out by applying the propagation law of errors, that contains terms of uncertainty of precision, recovery and standard purity. The expanded uncertainty is obtained applying a coverage factor K=2 (confidence level of 95%).
Results: The accuracy of the method can be considered good. Fat recoveries are obtained between 0,95 and 1,01 for the three concentration levels. The percentages of relative expanded uncertainty that are declared for each concentration level of fat in the samples of routines are the following ones: 0,50% (17,64%); 0,50-1,00% (13,09%); 1,00-16,55% (6,62%); 16,55-35,14% (6,00%); 35,14-50% (7,00%).
Conclusions: The estimated uncertainty is carried out by using the information generated during the proportional bias estimate process, and it can be applied to the future routine samples analysis by this analytic method.
Key words: Uncertainty. Recovery. Fat. Canned foods. Combat rations.
Introducción
La validación de un método analítico es un componente esencial de las medidas que el laboratorio debe tomar para obtener datos analíticos fiables y comparables, y su autenticidad depende de la determinación cuidadosa de los criterios de calidad del método analítico. En este sentido la responsabilidad de los analistas para asegurar que los informes proporcionan datos exactos tiene una gran importancia en las decisiones en salud pública que se puedan tomar en el ámbito de las Fuerzas Armadas.
Se admite generalmente que la adecuación a la finalidad o requerimientos perseguidos de un método analítico "fitness for purpose" no puede ser evaluado sin una estimación de la incertidumbre de la medida para comparar con la confianza requerida. Precisamente uno de los requisitos de la ISO 170251 para la acreditación es la estimación de la incertidumbre expandida, parámetro fundamental para la implementación de la trazabilidad como medio para asegurar la comparabilidad de resultados y una parte esencial de cualquier análisis cuantitativo. Puede suceder que dos métodos dan el mismo resultado, pero si la incertidumbre es demasiado alta muchos resultados podrían estar fuera de los límites de aceptabilidad.
Los principios establecidos en GUM2, publicada por ISO, también conocida como aproximación "bottom-up" para la evaluación y expresión de la incertidumbre en un amplio rango de medidas, son significativamente diferentes de los métodos utilizados en análisis nutricional, en los cuales generalmente se hace uso del método de forma global. Parámetros como la precisión y recuperación obtenidos durante la validación in-house constituyen una alternativa excelente para evaluar la incertidumbre de los métodos analíticos y evitar un excesivo coste en tiempo y esfuerzo3-7.
La amplia variabilidad de matrices y concentraciones de grasa de las conservas de raciones de combate, hace que los laboratorios de análisis nutricional tengan limitaciones en la validación de los métodos analíticos debido a la carencia de referencias con alto nivel de trazabilidad, tales como materiales certificados de referencia, ante la dificultad de emparejar la matriz y la concentración de las muestras de rutina con el material de referencia certificado. Por tanto, se recurre a referencias con un más bajo nivel de trazabilidad, como las muestras fortificadas o adicionadas, recurso menos costoso, con la desventaja de que el analito adicionado puede tener un comportamiento diferente del analito nativo, pero es una de las referencias frecuentemente utilizadas en los laboratorios de análisis.
La IUPAC "Gold book"8 define la recuperación como la fracción de la cantidad total de una sustancia recuperable siguiendo un procedimiento químico. El término recuperación se usa habitualmente para expresar la relación entre el analito encontrado con el método analítico frente a un valor de referencia. La IUPAC9 ha recomendado usar el término "recuperación aparente", que se define como el valor observado, x(obs), derivado de un procedimiento analítico por medio de un gráfico de calibración dividido por el valor de referencia, x(ref). La "recuperación aparente" puede estimarse con técnicas de regresión10 o mediante el método de la recuperación media, según la siguiente expresión, donde Rm, es la recuperación media; Rij es la recuperación obtenida para la muestra j adicionada con la cantidad i de analito; n es el número total de replicas adicionadas de analito, y p es el número de matrices adicionadas. Rij se calcula como la relación entre la concentración encontrada y adicionada (Ceij/Caij).

Uno de los parámetros más importantes en la validación de un método analítico es la estimación de la veracidad, que se considera en términos de recuperación11 e implica la estimación del sesgo proporcional, que varía con la concentración y se evalúa en términos de "recuperación aparente", variando la concentración adicionada y el intervalo de matrices de la muestras de rutina.
Objetivos
Validación in-house y estimación de la incertidumbre mediante ensayos de recuperación en la determinación de grasa en conservas de raciones de combate, utilizando la información generada en el proceso de validación, para poder aplicarse al análisis de futuras muestras de rutina con este método analítico.
Material y métodos
Preparación de la muestra
Las conservas de las raciones de combate se trituran en la trituradora (Vorwerk, Germany) aplicando varios períodos de 10 sg de duración a la intensidad máxima, con lo que se consigue una mezcla de consistencia pastosa. La temperatura de la muestra al final de la trituración no debe sobrepasar 24oC y se mide con un termómetro con certificado de calibración. El tamaño de partícula se comprueba sobre un tamiz ISO 3310-1:2000 con tamaño de la malla ≤ 1mm y diámetro de 200 mm.
Las muestras una vez homogeneizadas se analizan tan pronto como sea posible y siempre dentro de las 24 horas siguientes, manteniéndose en refrigeración a una temperatura 2-8o C. No obstante, es posible congelar las muestras a - 22oC, descongelarlas en refrigeración, y a continuación analizarlas.
Método de análisis
El método de análisis se ha modificado con respecto a la norma ISO 1443-197312 y a los métodos analíticos de la AOAC13, y se ha elaborado un procedimiento interno de análisis de grasa en conservas (animales, vegetales y platos preparados) de las raciones de combate. En la metodología de validación se utilizan tres matrices representativas de la variabilidad de las muestras de rutina o futuras (melocotón en almíbar, paté y sardinas en aceite). A las muestras se les añade una referencia estándar (aceite de ricino, riqueza 99,7%) (Sigma-Aldrich) con lo que se obtienen concentraciones de grasa en el centro y en los extremos del rango de trabajo. Las muestras adicionadas con el analito y las muestras sin adicionar se analizan en la misma serie, en condiciones de precisión intermedia, y mediante un diseño anidado14, se varían dentro del laboratorio aquellos factores que afectan al procedimiento analítico (cantidad adicionada, matriz, precisión intermedia), hallándose la recuperación por triplicado.
La determinación de la grasa se realiza por gravimetría, previa hidrólisis con ácido clorhídrico 3N en ebullición durante 1 hora. La extracción de la grasa se realiza con éter de petróleo 40-60o mediante el sistema Soxtec System Tecator.
El cálculo de la repetibilidad se realiza a partir de la expresión siguiente, donde Sr es la estimación de la desviación estándar de la repetibilidad de las recuperaciones y n el número de series. Se asume que el error sistemático es el mismo en el intervalo de aplicación del método.

El límite de repetibilidad se obtiene según la expresión: 1,96  , para un nivel de confianza del 95%.
, para un nivel de confianza del 95%.
Para el estudio del sesgo proporcional del método analítico se calcula la recuperación media, Rm, y el estadístico t, de dos colas, para determinar si el valor medio obtenido en las recuperaciones de grasa es significativamente diferente de 115. La expresión siguiente se utiliza para hallar la recuperación aparente de cada serie:

En la que Ca+m= Cantidad añadida de analito y cantidad de analito en la muestra, Cm= Cantidad de analito en la muestra, y Ca= Cantidad de analito añadida a la muestra de análisis.
Y si Rm, que es una estimación media experimental de la recuperación aparente, obtenida con la expresión (1) no es estadísticamente diferente de 1, se debe satisfacer la siguiente inecuación:

donde, Rm es la recuperación media, u(sesgo) es la incertidumbre del sesgo, y t/2,eff es el valor tabulado de dos colas para los grados de libertad efectivos asociados a u(sesgo) al 95% de confianza, calculados utilizando al aproximación de Welch-Sattherthwaite. La incertidumbre del sesgo se estima según la expresión:

donde, s es la desviación estándar de las n recuperaciones, y n el número de recuperaciones.
Fuentes de incertidumbre
Las fuentes de incertidumbre del mensurando se pueden identificar construyendo un diagrama causa-efecto. Entre las más significativas tenemos la masa de la muestra y la masa de la grasa extraída, la pureza del material estándar, variabilidad de las muestras de rutina, repetibilidad de la recuperación, pobre representación entre matrices utilizadas en el experimento y las correspondientes a las muestras de rutina y pobre representación del analito nativo en relación al analito adicionado. Este procedimiento resulta ortodoxo y la cuantificación de la incertidumbre estándar en etapas intermedias no siempre es fácil. En consecuencia se decide realizar la estimación de la incertidumbre de una forma global, considerando el proceso analítico de forma conjunta, y siguiendo las recomendaciones de la guía EURACHEM/CITAC16, se aprovecha la validación del método para calcular la incertidumbre de los resultados, de tal manera que se agrupan los términos relativos incertidumbre de la precisión, incertidumbre de la recuperación e incertidumbre de la pureza del material estándar que se adiciona a la muestra.
La incertidumbre combinada estándar uc(y), usualmente denominada ley de propagación de la incertidumbre17, basada en una serie de primer orden de Taylor, y asumiendo que todas las fuentes de incertidumbre son proporcionales a la concentración de analito, los componentes individuales para modelos que implican un cociente y= p/(q x r x...), se expresa por la suma de incertidumbres relativas (u(p)/p) de los parámetros independientes, expresados como desviación estándar relativa:

Y la incertidumbre expandida (U), se obtiene multiplicando la incertidumbre combinada estándar (uc), por un factor de cobertura K=2, asimilable a un nivel de confianza del 95%.
Resultados
En la tabla 1 se exponen los resultados estimados de las recuperaciones de grasa en matrices representativas de las muestras de rutina para los tres niveles de concentración.

Se obtiene un valor texp < tα/2,eff, y por consiguiente, Rm no difiere estadísticamente de 1, dentro de los límites de su incertidumbre, y se verifica la ausencia de sesgo proporcional significativo del método. No obstante, para niveles bajos de concentración de grasa se halla un sesgo proporcional significativo. En la tabla 2 se muestran los datos de repetibilidad y límite de repetibilidad para porcentajes de grasa bajo, medio y alto.

La diferencia máxima entre los resultados de dos recuperaciones utilizando muestras fortificadas, de acuerdo con la sistemática de validación, realizadas por el mismo analista, será en tanto por uno, igual o inferior, a los datos de la Tabla 3.

Asimismo, la exactitud del método se considera buena, obteniéndose recuperaciones entre 0,90 y 1,13, con unas recuperaciones medias entre 0,95 y 1,01, incluyendo los tres niveles de concentración.
La precisión intermedia del método o reproductibilidad intra-laboratorio, estimada sobre los datos de recuperación, expresada como desviación estándar relativa (DER), se considera satisfactoria, según los siguientes porcentajes:
- Nivel de concentración bajo: 4,05%
- Nivel de concentración medio: 2,17%
- Nivel de concentración alto: 5,64%.
Y se estima el intervalo de recuperaciones válido, entre 0,86 y 1,16 (nivel de confianza del 95%), para los tres niveles de concentración estudiados. En las tablas 4, 5 y 6 se exponen los datos con los parámetros utilizados para la estimación de la incertidumbre expandida relativa (Uexp rel).
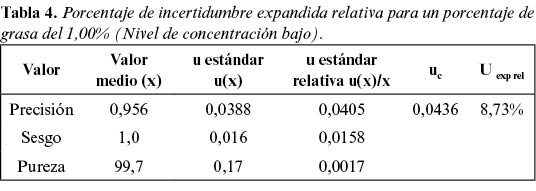


El límite de cuantificación del método se estima en un 0,2% y la estimación de la incertidumbre expandida relativa, Uexp rel, para la concentración del 0,50% de grasa, se realiza a partir de la incertidumbre absoluta constante para la concentración de 1, 00%, y se obtiene un valor del 17,46%.
La incertidumbre expandida relativa (Uexp rel) para cada nivel de concentración de grasa son las que se declaran en la Tabla 7.

En la figura 1 se representa la contribución de la precisión, sesgo y pureza del material estándar a la contribución de la incertidumbre expandida relativa combinada.

Discusión
El procedimiento analítico ha sido validado globalmente con inclusión del tratamiento de la muestra antes del análisis, y abarca todo el rango de concentraciones del analito, que representan el rango normal de trabajo del método o la concentración esperada en la muestra, y las matrices representativas de las muestras de rutina o futuras. En todo caso se ha considerado un equilibrio entre coste y beneficio en la estimación de la incertidumbre, dada la gran variabilidad de matrices que pueden afectar a las muestras de rutina.
Las muestras fortificadas o adicionadas se preparan añadiendo aproximadamente la misma cantidad de analito a cada muestra individual, con concentración inicial de analito, por considerarse poco práctico realizar submuestreos a partir de una masa de muestra adicionada, y con ello se evita introducir una incertidumbre derivada de la homogeneización.
En aquellos casos en los que la concentración de analito nativo es apreciable, la adición del analito se realiza de tal manera que la concentración final en la porción test sea similar a la concentración de analito nativo para evitar incurrir en una elevada incertidumbre relativa en la recuperación del analito adicionado18. Es de esperar que el resultado resultante de la estimación de la recuperación en muestras reales tenga un sesgo, ya que una incompleta recuperación deriva a sesgo, sobretodo en métodos analíticos de estas características que incluyen varios pasos de transferencia de masa que implican eficiencias diferentes del 100% (extracción, digestión, etc.)19.
La recuperación del analito adicionado puede diferir de la recuperación del analito nativo, ya que no se produce una total destrucción de la muestra durante la hidrólisis, además las muestras adicionadas poseen matrices complejas sólidas o semisólidas, y tampoco se espera que el analito sea quimioabsorbido por la matriz, y el analito adicionado podría tener una recuperación más alta que el analito nativo al tener distinto comportamiento. Por este motivo, se obtienen recuperaciones inferiores y superiores al 100% en cuanto a "recuperación aparente", debido a que en el cálculo de la misma se introducen errores procedentes de la cantidad de grasa de la muestra con analito nativo y de los procedentes de la extracción de la grasa adicionada. Es de resaltar que la veracidad de los métodos analíticos no puede ser garantizada por la posibilidad de existencia de un sesgo constante20, independiente de la concentración, cuyo estudio será objeto de futuras investigaciones. Por otro lado EURACHEM y otras aproximaciones no consideran el hecho de que pueda existir un sesgo constante en los métodos analíticos.
Una idea más aproximada del comportamiento del analito nativo sería el análisis de un MRC que contenga un analito más difícil de extraer que el de las muestras reales, y la recuperación observada proporcionará una recuperación "worst case" y se podría asumir que la recuperación de las muestras reales probablemente estarán más próximas a 1.
Siguiendo las recomendaciones de la IUPAC21 la adición se hace a tres niveles de concentración (bajo, medio, alto), en los extremos y en el valor medio del intervalo de concentraciones de las muestras de rutina o muestras futuras, ya que la recuperación y su incertidumbre estándar suelen depender de la concentración de analito22, y el cambio de la incertidumbre con la concentración es atribuible a la recuperación del método23.
En este trabajo experimental se utiliza el método de la recuperación media, que es el resultado de la estimación de la recuperación de cada cantidad de analito adicionado a diferentes matrices que son representativas de las muestras futuras. Este método podría proporcionar incertidumbres menores que el método de adiciones estándar. Precisamente una de las limitaciones de la técnica de adiciones estándar es que puede obtenerse una estimación incorrecta del sesgo proporcional si se produce un efecto interactivo entre la matriz y el analito24.
Las consideraciones estadísticas recomiendan realizar 3≤ n ≤5 replicados en cada nivel de concentración y, en este caso, el empleo de un diseño anidado de tres factores (cantidad adicionada, factor analito-matriz y precisión intermedia), permite obtener información útil para la estimación de la incertidumbre25, ya que los resultados de los ensayos de recuperación se ven afectados por estos factores.
De acuerdo con las recomendaciones anteriores, se realizan análisis por triplicado, que además de ajustarse a las características del instrumento, permite analizar conjuntamente las muestras con analito nativo y analito adicionado. El número de replicados realizados se considera suficiente para proporcionar una fiable estimación de la desviación estándar de los resultados. Estadísticamente se ha demostrado que con menos de 6 replicados el intervalo de confianza de la población llega a ser muy amplio, y más allá de 15 replicados hay pocos cambios en el intervalo de confianza.
En la bibliografía se citan datos de recuperación con un rango de tolerancia para residuos químicos entre el 70-120%26. Similares datos se han obtenido en recuperaciones de grasa en alimentos diversos (77-134%)27. En otros trabajos se detallan unas recuperaciones aceptables, en%, dependiendo del nivel de concentración de analito: 100% (98-102); 10% (98-102); 1 (97-103)28. En nuestro caso se observa una ligera desviación respecto a estos datos para concentraciones de grasa del 1%, al obtenerse una recuperación media del 95%.
En 3 ensayos de aptitud realizados en nuestro laboratorio, para un valor de grasa asignado del 2,66%, se ha conseguido una recuperación del 92%; para un valor del 30,40%, se recupera un 97,07%, y para un valor del 18,26%, se recupera un 95,70%. Por consiguiente, existe un sesgo de recuperación que puede depender, especialmente de la concentración, y de la matriz, de errores aleatorios, y sería factible corregir los resultados para niveles de concentración bajos grasa.
Otro aspecto importante de la validación es el estudio de resultados anómalos "outliers", que se realiza mediante el criterio R de Grubb´s, recomendado por la (ISO 5725-2)29, que supone que la población es normal. En este sentido el teorema del límite central proporciona el fundamento estadístico para esperar una distribución normal en una media de datos experimentales sujetos a errores aleatorios. No obstante, para verificar que los datos individuales se ajustan a una distribución normal se ha utilizado el test de Kolgomorov-Smirnov, test de asimetría, test de curtosis y el diagrama de puntos. Los resultados obtenidos con el test de curtosis (momento de cuarto orden) y asimetría (momento de tercer orden) para una distribución normal se hallan dentro del intervalo [-2, 2].
Aunque existen muchos tests estadísticos para comprobar la normalidad de los datos, y ninguno garantizado para rechazar o conservar los valores de datos individuales, sólo unos pocos tienen la suficiente potencia para ser usados en análisis nutricional, en donde habitualmente se manejan pocos datos. Tampoco es infrecuente encontrar resultados contradictorios en contrastes de datos anómalos, por ejemplo, entre el contraste de Grubb´s y el test Q de Dixon. No cabe duda, que al rechazar un valor la precisión suele mejorar, de ahí la importancia de ser cauto a la hora de rechazar datos anómalos, y sobre todo cuando las medidas se registran pocas veces, el rechazo de un valor origina una gran variación sobre la media y la desviación estándar. Esta dificultad explica el uso creciente de métodos estadísticos no paramétricos y robustos, al ser insensibles a valores extremos. No obstante, nuestro criterio es buscar las causas que generan datos anómalos, incluyendo factores humanos30, antes de proceder a descartarlos. En todo caso los criterios subjetivos no son válidos para considerar un resultado como discrepante.
Cuando la recuperación aparente no es estadísticamente diferente de 1, se puede concluir que el método analítico no tiene sesgo proporcional significativo. Pero cuando se valora la veracidad siempre existe una probabilidad de concluir que el sesgo proporcional no es significativo. Los resultados de simulación de la recuperación aparente con el método Monte-Carlo muestran que el sesgo no significativo puede infraestimar la incertidumbre de los resultados y los porcentajes de exactitud de futuros resultados serían más bajos del 95%.
En realidad no sería necesario aplicar un factor de recuperación Xcorr= FHom x c/Rm a las futuras muestras de rutina que se analizarán con el procedimiento31-34, salvo para niveles de concentración ≤1% de grasa en donde la recuperación es significativamente diferente de 1. En la expresión anterior, c es la concentración al analizar la muestra, Rm la recuperación media, Xcorr, resultado obtenido para futuras muestras y Fhom será 1 a efectos prácticos.
La precisión del método constituye un factor importante en la incertidumbre combinada, expresada como desviación estándar de las recuperaciones, y es similar en orden de magnitud para los tres niveles de concentración estudiados. Esto indica que esta desviación estándar es independiente de la concentración. Cabe decir que la relación entre la precisión, expresada como desviación estándar, y la concentración en un sistema analítico puede seguir otro tipo modelos, incluyendo modelos hiperbólicos35.
Los niveles de precisión, expresados como desviación estándar relativa, hallados experimentalmente, en tres niveles de concentración son los siguientes: nivel bajo (1,00%/2,20%), nivel medio (16,55%/1,88%) y nivel alto (35,14%/1,14%), son ligeramente inferiores a los que predice la ecuación de Horwitz de acuerdo con la ecuación empírica:

Los ensayos de recuperación puede verse afectados por otros componentes que pueden afectar a la recuperación40, como por ejemplo, el factor Rs, que afecta a las diferencias de recuperación para una muestra real comparada con la recuperación observada para el material usado para estimar Rm , que es una estimación de la recuperación media del método. Por este motivo se analizan un rango representativo de muestras adicionadas, cubriendo matrices típicas y concentraciones de analito. Otro factor a considerar es, Rrep, que afecta a la muestra adicionada y puede tener un comportamiento diferente a la muestra real con analito nativo. Por lo tanto, se deduce que la incertidumbre en R, u(R), tendrían contribuciones de u (Rm), u (Rs), y u (Rrep). La incertidumbre u(Rs) es, por tanto, la desviación estándar de las recuperaciones medias para cada muestra tipo, y se ha considerado despreciable, así como u(Rrep).
Hay que tener presente que la incertidumbre de la medida a muy baja concentración, por ejemplo, para porcentajes de grasa del 0,5%, cercana al límite de detección del método, que se estima en un 0,2%, podría incrementarse fuertemente, y la suposición de que la incertidumbre relativa es constante no puede aceptarse en algunos casos. Por ello, se aplica una transición a incertidumbre constante absoluta en el más bajo límite del rango de aplicación, es decir, una incertidumbre absoluta por debajo del 1%, y en el intervalo del 0,50% al 1% de grasa, se obtiene una incertidumbre relativa estimada del 13,09%.
Existe un conocimiento bien estabilizado entre la incertidumbre y la concentración, que puede ser usado para justificar la conversión de la incertidumbre absoluta en un valor relativo. La incertidumbre expandida relativa no se espera que varíe como una función de la concentración, cuando las medidas se han hecho por encima de su límite de detección (> 10 veces), y la incertidumbre absoluta cambiaría proporcionalmente con la concentración en cada serie futura41.
Además, el parámetro que más contribuye a la medida de la incertidumbre es la precisión del método, y probablemente se explica por la complejidad de la matriz y los errores aleatorios. Es de reseñar que la contribución al resultado final debido a la incertidumbre de la cantidad de material estándar adicionado se considera despreciable.
La pureza de la referencia estándar (aceite de ricino) se ha considerado un parámetro de influencia en la determinación de la incertidumbre pese a tener una contribución menor del 20% de la mayor contribución de la incertidumbre combinada estándar relativa. En este caso para el estándar la pureza es ≥99,7%, y se toma el intervalo entre el 99,7% y el 100% como límite de una función distribución de probabilidad que se asume rectangular42,43 (media ± 58%), con una anchura de 2a del 0,3%, que corresponde a una incertidumbre estándar de 0,6 x 0,3%= 0,0018.
La incertidumbre expandida, para k=2, se estima en un 10,56%, considerando globalmente los tres niveles de concentración, y resulta inferior a la estimación (19,15%), utilizando los datos de reproducibilidad de tres estudios interlaboratoriales citados anteriormente.
Conclusiones
1. La incertidumbre estimada para cada nivel de concentración se realiza utilizando la información generada durante el proceso de validación y puede aplicarse al análisis de futuras muestras de rutina con este método analítico.
2. La incertidumbre asociada con la precisión es el término que más contribuye a la incertidumbre combinada.
3. Se observa que la recuperación depende de la concentración de analito.
Bibliografía
1. ISO/IEC 17025. General requirements for the competence of testing and calibration laboratories. European Committee for Standard. Brusells. 2005. [ Links ]
2. Guide to the Expression of Uncertainty in Measurement, ISO, Geneva, Switzerland, 1993. [ Links ]
3. Ellison SLR, Barwick VJ. Using validation data for ISO measurement uncertainty estimation. Part 1. Principles of an approach using cause and effect analysis. Analyst 1998; 123: 1387-1392. [ Links ]
4. Maroto A, Boqué R, Riu J, Rius X. Effect of non-significant proportional bias in the final measurement uncertainty. Analyst 2003; 128:373-378. [ Links ]
5. Feinberg M, Laurentie M. A global approach to method validation and measurement uncertainty. Accred Qual Assur 2006; 11: 3-9. [ Links ]
6. Cuadros Rodríguez L, Torres Hernández ME, Almansa López E, Egea González FJ, Arrebola Liébanas FJ, Martinez Vidal JL. Assessment of uncertainty in pesticide multiresidue analytical methods: main sources and estimation. Anal Chim Acta 2002; 454:297-314. [ Links ]
7. Williams A. Introduction to measurement uncertainty in chemical analysis. Accred Qual Assur 1998; 3: 92-94. [ Links ]
8. IUPAC "Gold bood". Internet: http://goldbook.iupac.org. Último acceso: 4/02/2013. [ Links ]
9. Thompson M, Ellison S.L.R., Wood R.A. Harmonised Guidelines for single-laboratory validation of methods of analysis. Pure Appl Chem 2002; 74 (5): 2201-2205. [ Links ]
10. Maroto A, Boqué R, Riu J, Rius FX. Measurement uncertainty in analytical methods in which trueness is assessed from recovery assays. Anal Chim Acta 2001; 440:171-184. [ Links ]
11. Barwick VJ, Ellison LR. VAM Project 3.2.1. Development and Harmonization of Measurement Uncertainty Principles. Part D: Protocol for Uncertainty Evaluation from Validation Data, January 2000. Report No: LGC/VAM/1998/088. [ Links ]
12. ISO 1443-1973. Meat and Meat Products. Determination of total fat content. [ Links ]
13. McNeal JE. Meat and Meat Products. En: Official Methods of Analysis. Volume two. AOAC. 15th Edition. USA: 1990: 931-948. [ Links ]
14. ISO Guide 5725-3:1994/Cor.1:2001. Accuracy (Trueness and Precision) of Measurement Methods and Results. Part 3: Intermediate measures of the precision of a standard measurement method. Genève. [ Links ]
15. Bagur G, Sánchez-Viñas M, Gázquez D, Ortega M, Romero R. Estimation of the uncertainty associated with the standard addition methodology when a matriz effect in detected. Talanta 2005; 66: 1168-1174. [ Links ]
16. Ellison SLR, Rosslein M, Williams A. EURACHEM/CITAC Guide. Quantifying Uncertainty in Analytical Measurement. 2a Edition. 2000. [ Links ]
17. Mekid S, Vaja D. Propagation of uncertainty: Expressions of second and third order uncertainty with third and fourth moments. Measurement 2008: 41: 600-609. [ Links ]
18. Thompson M, Ellison SLR, Fajgeli A, Willetts P, Wood R. Harmonised guidelines for the use of recovery information in analytical measurement. Pure Appl Chem 1999; 71 (2): 337-348. [ Links ]
19. Bettencourt da Silva RJN, Lino MJ, Santos RJ, Camoes FGFC. Estimation of precision and efficiency mass transfer steps for the determination of pesticides in vegetables aiming at the expression of results with reliable uncertainty. Analyst 2000; 125: 1459-1464. [ Links ]
20. Hund E, Massart DL, Smeyers-Verbeke J. Operacional definition of uncertainty. Trends Anal Chem, 2001; 20 (8): 394-406. [ Links ]
21. Thompson M, Ellison S.L.R., Wood R.A. Harmonised Guidelines for single-laboratory validation of methods of analysis. Pure Appl Chem 2002; 74 (5): 835-855. [ Links ]
22. Gustavo González A, Herrador MA, Asuero AG. Practical digest for evaluation the uncertainty of analytical assays from validation data according to the LGC/VAM protocol. Talanta 2005; 65: 1022-1030. [ Links ]
23. Armishaw P. Estimating measurement uncertainty in a afthernoon. A case study in the practical application of measurement uncertainty. Accred Qual Assur 2003; 8:218-224. [ Links ]
24. García Campaña AM, Rodríguez Cuadros L, Muñoz Aybar J, Alés Barbero F. Chemometric protocol to validate an analytical method in the presence of corrigible constant and proportional systematic errors. J AOAC International 1997; 80: 657-664. [ Links ]
25. Willetts P, Wood R. Measurement uncertainty - a reliable concept in food analysis and for the use of recovery data? Accred Qual Assur 1998; 3:231-236. [ Links ]
26. Parker GA. Validation of methods used in the Florida Department of Agricultural and Consumer Services Chemical Residue Laboratory. J Assoc Off Anal Chem 1991; 74: 868-871. [ Links ]
27. Thiex NJ, Gildemeister B. Crude fat, hexanes extraction, in feed, cereal grain, and forage (Randall/Soxtec/Submersion Method): Collaborative Study. J AOAC International 2003; 86:899-908. [ Links ]
28. González AG, Herrador MA. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends Anal Chem 2007; 26:227-238. [ Links ]
29. ISO Guide 5725-2:1994/Cor.1:2001. Accuracy (Trueness and Precision) of Measurement Methods and Results. Part 2: Intermediate measures of the precision of a standard measurement method. Genève. [ Links ]
30. Kuselman H, Pennechi F, Fajgelj A, Karpov Y. Human errors and reliability of test results in analytical chemistry. Accred Qual Assur 2013; 18: 3-9. [ Links ]
31. Linsinger TPJ. Use of recovery and bias information in analytical chemistry and estimation of its uncertainty contribution. Trends Anal Chem 2008; 27:916-923. [ Links ]
32. Haesselbarth W. Accounting for bias in measurement uncertainty estimation. Accred Qual Assur 2004; 9: 509-514. [ Links ]
33. Synek V. Effect of significant bias and its uncertainty on the coverage probability or uncertainty intervals. Part 2. Evaluation for a found insignificant experimental bias. Talanta 2007; 70: 1304-1311. [ Links ]
34. Synek V. Effect of significant bias and its uncertainty on the coverage probability or uncertainty intervals. Part 1. Evaluation for given value of true bias. Talanta 2006; 70: 1024-1034. [ Links ]
35. Thompson M. Variation of precision with concentration in an analytical system. Analyst 1988; 113: 1579-1587. [ Links ]
36. Thompson M, Lowthan PJ. The Horwitz Function revisited. J AOAC International 1997; 80: 676-679. [ Links ]
37. Alder L, Korth W, Patey AL, Van der Schee H. Estimation of measurement uncertainty in pesticide residue analysis. J AOAC International 2001; 84: 1569-1574. [ Links ]
38. Lisinger TPJ, Ralf DJ. Limitations of the application of the Horwitz equation. Trends Anal Chem 2006; 25: 1125-1130. [ Links ]
39. Wood R. How validate analytical methods. Trends Anal Chem 1999; 18:624-632. [ Links ]
40. Barwick VJ, Ellison SLR. Measurement uncertainty: Approaches to the evaluation of uncertainties associated with recovery. Analyst 1999; 124:981-990. [ Links ]
41. Ramsey MH, Ellison SLR. Reply to comments on EURACHEM /CITAC guide "Measurement uncertainty arising from sampling". Accred Qual Assur 2010; 15: 533-535. [ Links ]
42. Wong KS. Purity verification and measurement uncertainty. Accred Qual Assur 2010; 15: 337-341. [ Links ]
43. BIPM, IEC, IFCC, ISO, IUPAC, IUPAP, OIML. Guide to the expression of uncertainty in measurement ISO, Genève, 1993. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Roberto Pérez Grana.
Centro Militar de Veterinaria de la Defensa.
Servicio de Bromatología y Seguridad Alimentaria.
C/ Darío Gazapo, 3.
28024. Madrid.
Tfno. 91-5122515. rpergra@oc.mde.es
Recibido: 24 de junio de 2013
Aceptado: 23 de enero de 2014

