










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.8 no.1 Madrid ene./mar. 2016
Classic non-deforming osteogenesis imperfecta. Report of a new mutation in the COL1A1 gene in two cases in the same family
Osteogénesis imperfecta forma clásica no deformante. Comunicación de una nueva mutación en el gen COL1A1 en dos casos de la misma familia
Pavón de Paz I.1, Gil Fournier B.2, Navea Aguilera C.1, Ramiro León M.S.2, Modroño Móstoles N.1 and Guijarro de Armas G.1
1 Servicio de Endocrinología y Nutrición
2 Servicio de Genética
Hospital Universitario de Getafe - Madrid (España)
SUMMARY
Osteogenesis imperfecta (OI), is a rare condition which is heterogeneous in clinical and genetic terms. Several types have been described and its main feature is bone fragility. It is generally caused by gene mutations in those genes which codify for the α1 and α2 of the type 1 collagen (COL1A1 and COL1A2) with dominant autosomal heredity.
We report the case of two relatives (father and daughter) with OI whose genetic study shows a mutation in COL1A1 previously undetected: the deletion of a Guanine, G(c.3524delG). Clinical aspects, heredity and reproductive options of the patients affected are considered.
Key words: osteogenesis imperfecta, genetic research, COL1A1 gene.
RESUMEN
La osteogénesis imperfecta (OI), es una patología poco frecuente y muy heterogénea desde el punto de vista clínico y genético. Su característica principal es la fragilidad ósea, habiéndose descrito varios tipos.
Generalmente es causada por mutaciones en los genes que codifican para las cadenas α1 y α2 del pro-colágeno tipo 1 (COL1A1 y COL1A2) con herencia autosómica dominante.
Comunicamos los casos de dos pacientes (padre e hija) con OI cuyo estudio genético muestra una mutación en COL1A1 no conocida previamente: la deleción de una Guanina, G(c.3524delG). Se repasan aspectos clínicos, de herencia y opciones reproductivas de los pacientes afectados.
Palabras clave: osteogénesis imperfecta, estudio genético, gen COL1A1.
Introduction
Osteogenesis imperfecta (OI), also known as "brittle bone disease", is a rare and very heterogeneous disease from the clinical and genetic point of view. It is due to mutation of genes involved in the formation of type 1 collagen, affects one in every 15,000-20,000 live births. Although its main, common characteristic is bone fragility, several types have been described depending on genetic radiological and clinical features [1-3].
Generally, OI is caused by heterozygous mutations in the genes encoding the α2 and α1 chain of type 1 procollagen (COL1A1 and COL1A2 genes) but other genes have been identified. Mutations in the COL1A1 and COL1A2 genes are inherited in an autosomal dominant pattern [1-4].
We report two cases of two separate patients (father and daughter) affected by a non-deforming type of OI (probably Sillence type 1) in which the genetic study shows a mutation in the COL1A1 gene not previously known. This heterozygous deletion of a guanine, G (c.3524delG), is not described in the literature or in the databases.
Case 1
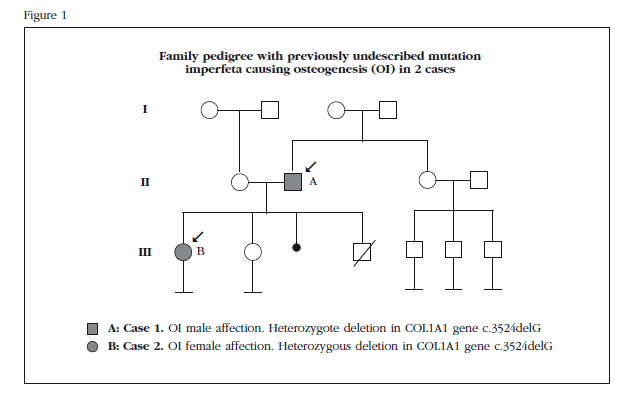
A 65-year-old male patient diagnosed with OI following a study that showed the daughter was also affected. He showed no previous family history of the disease (Figure 1).
He reported a history of multiple fractures (approximately 6-7) during childhood and adolescence after minor trauma, the first of the humerus at 2 years old. In adulthood he had two new fractures in elbow and shoulder. Both had been treated conservatively. He was treated with calcium during childhood. He had also presented throughout his life several multiple sprains and muscle tears. Diagnosed with otosclerosis, he had undergone stapedectomy surgery of both ears.
He was referred to our clinic for osteoporosis detected in bone densitometry (DXA), which showed a T-scores of -3.4 at the lumbar spine (L1-L4), femoral neck -3 and -2.8 in total femur. It was asymptomatic.
On physical examination, the size of 162 cm was noteworthy as was blue sclera and the presence of dentinogenesis imperfecta. He did not present thoracic, lumbar vertebral column or member deformities, except in the right elbow (post-fracture). No hypermobility was detected.
The study of calcium-phosphorus metabolism showed normal levels of calcium, phosphorus, urinary calcium, and parathyroid hormone (iPTH). Levels of 25-hydroxyvitamin D (25OHCC) were insufficient: 22 ng/ml (desirable values >30 ng/ml). The bone turnover markers were in normal range. other endocrine causes of osteoporosis were excluded.
As part of our study protocol of patients suffering from OI, an X-ray of cervical spine basilar impression was taken and a chest image in which degenerative changes were evident in the column. ECG showed minimal dilatation of the ascending aorta; respiratory function tests were normal, and abdominal ultrasound ruled out nephrolithiasis.
Genetic study was carried out using massive sequencing by NGS (Next-Generation Sequencing) of the COL1A1, COL1A2, and LEPRE1 CRTAP genes, detected in the COL1A1 gene deletion of a guanine heterozygosity (c.3524delG).
This mutation results in a change in the reading frame, which, at the level of collagen protein, results in a premature stop codon (p.Gly1175Valfs*64) so it is likely to be a pathogenic change. Other detected changes were considered polymorphisms.
Weekly treatment was recommended with alendronate along with daily supplements of calcium and vitamin D, showing a slight densitometric improvement after 1 year of treatment (T-scores of -3.2 at the lumbar spine, femoral neck -2.9 and -2, 4 total femur).
Case 2
A 30-year-old patient, daughter of former patient (Figure 1), diagnosed in childhood with OI following a displaced fracture of tibia followed by broken collarbone after minor trauma. She then presented three new fractures, the most recent at age 12, which required surgical treatment (olecranon). Among other relevant history, multiple ankle sprains and right ear stapedectomy were highlighted. She had never been treated for this disease and presented asymptomatic.
The patient requested reproductive data regarding her chances of having a child free of disease.
Physical examination revealed a size of 153 cm, blue sclera and normal teeth. No deformities were observed at any level.
Laboratory tests were normal, except 25OHCC levels: 23 ng/ml, and the rest of the study (cervical spine radiography, echocardiography, spirometry, abdominal ultrasound). The results of densitometry showed normal BMD: T-scores of -0.9 at the lumbar spine (L1-L4), femoral neck and 0.0 -0.1 total femur. Vitamin D supplements were recommended.
Directed genetic study was performed to search for the identified mutation in her father, confirming that the child carries the same deletion in the COL1A1 gene heterozygosity as her father. Genetic counseling was conducted to report on the possible consequences for affection to offspring, the results of a genetic study and their advantages and risks, and inform them of the possible alternatives derived from the analysis.
Discussion
Type 1 collagen is a structural protein that forms part of the bone, skin, teeth, tendons, ligaments and sclera. In general, OI is caused by heterozygous mutations in the genes encoding the α1 chains and α2 type 1 (COL1A1 and COL1A2 genes) procollagen but also have identified other genes involved in the processing of type 1 collagen, such as CRTAP and LEPRE. The COL1A1 gene is located on chromosome 17 q21 region in-q22, COL1A2 gene and chromosome 7 in the region q22. Mutations in the COL1A1 and COL1A2 genes are inherited in a dominant autosomal pattern [1-4]. That is, every time a parental affection conceives a child, there is a 50% chance in every pregnancy of passing on the disease, regardless of sex.
Clinical abnormalities of OI related to COL1A1 and COL1A2 genes are primarily fractures without trauma or after minimal trauma, variable dentinogenesis imperfecta and hearing loss in adulthood. The severity of the clinical presentation depends on the effect of the mutation. Mutations that bring about a reduction in the amount of synthesized collagen forms are milder than those affecting the proteic structure [5].
There is a continuum from the most severe form, the perinatal-lethal (type 2 Sillence classification), individuals with severe deformities, short stature and impaired mobility (types III and IV Sillence) to patients virtually asymptomatic of dentinogenesis imperfecta normal height and predisposition to fractures, but with normal life expectancy (type I Sillence) [5].
The diagnosis of OI should be based on family history, history of fractures, usually spontaneous or with minimal trauma, short stature, sometimes associated with more or less severe deformities, and the presence of other clinical data such as blue or gray sclera, dentinogenesis imperfecta, ligamentous laxity and progressive hearing loss after puberty. Radiographic findings include osteopenia or osteoporosis, wormian presence of bones, skeletal deformities and fractures or its aftermath. A molecular genetic study is recommended to confirm the diagnosis [6-7].
The two cases described here show a mild form, the most frequent, with common characteristics such as predisposition to fractures with minimal trauma, short stature, blue sclera, ligamentous laxity prone to sprains and dislocations and hearing loss transmission mild form of early OI, but do not share other clinical data such as dentinogenesis imperfecta. In mild forms, it is possible to find a bone mineral density within normal limits because the DXA measures bone mineral and not collagen content [9]. It has been reported that there is no clear genotype-phenotype correlation even within the same family [6].
Genetic studies of genes COL1A1 and COL1A2 detect abnormalities in 90% of individuals with OI types I, II, III and IV of Sillence. Its sensitivity is similar to the quantitative and structural analysis of type 1 collagen in cultured fibroblasts obtained from a skin biopsy [8].
Mutations were found more often in the COL1A1 gene (up to 70% of cases) the COL1A2 gene in both cases and inherited autosomal dominant or behave as de novo mutations. In all, more than 1,500 different mutations have been described. In our patients, the study found a heterozygous deletion of a guanine (c.3524delG) in the COL1A1 gene. This mutation, not previously described in the literature, gives rise to a change in the interpretive framework, which level collagen protein, results in a premature stop codon (p.Gly1175Valfs*64), so it is very likely a pathogenic change. Other detected changes were considered polymorphisms without clinical association.
De novo mutations constitute 100% of cases with lethal perinatal-(type II Sillence), almost 100% in progressively deforming shapes and about 60% of non-classical deformities [6]. For the family history of our patients, the father suffered a de novo mutation that was conveyed to her daughter with autosomal dominant pattern. Knowing the form of inheritance, we proceeded to inform the patient and make genetic counseling, including discussion of potential risks and potential reproductive choices.
The daughter (Case 2) inquired as to how to have a child free of this disease. Because the mutation that causes the OI has identified there are 3 reproductive options: PGD, following treatment of in vitro fertilization, which allows genetically tested embryos and select non affections of OI to be transferred to uterus; another option would be an IVF treatment with donor eggs. This technique avoids the gestation of a child with OI, because it replaces the parent's diseased affected gamete by an anonymous healthy gamete. Another option would be to conceive a child and prenatal diagnosis after obtaining fetal cells by chorion biopsy or genetic amniocentesis, on which can be directed genetic studies of OI (only when the responsible disease mutation is already known in the family, which will be searched for in the fetus) [10].
The treatment of choice in adult patients with OI is not clearly established, but several studies have demonstrated the efficacy of both oral bisphosphonates as intravenous [11,12]. The usefulness of other drugs such as denosumab [13] and parathyroid hormone [14] has yet to be recognized.
![]() Correspondence:
Correspondence:
Isabel Pavón de Paz
Servicio de Endocrinología
Hospital Universitario de Getafe
Ctra. de Toledo, Km 12,5
28905 Getafe - Madrid (España)
Correo electrónico: pavonisa@yahoo.es
Date of receipt: 26/01/2016
Date of acceptance: 16/03/2016
Bibliography
1. Marini JC. Osteogenesis imperfecta: comprehensive management. Adv Pediatr 1988;35:391-6. [ Links ]
2. Plotkin H. Syndromes with congenital brittle bones.BMC Pediatr 2004;4:16-21. [ Links ]
3. Prockop DJ, Kivirikko KI. Heritable diseases of collagen. N Engl J Med 1984;31:376-86. [ Links ]
4. Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans. Acta Biochim Pol 2002;49:433-41. [ Links ]
5. Van Dijk F, Sillence D. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014;164:1470-81. [ Links ]
6. Steiner RD, Adsit J, Basel D. COL1A1/2-Related Osteogenesis Imperfecta. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® (Internet). Seattle (WA): University of Washington, Seattle; 1993-2015. [ Links ]
7. Van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, Rohrbach M, et al. EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta. Eur J Hum Genet 2012;20:11-9. [ Links ]
8. Van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PGJ, van Rijn RR, et al. Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol 2011;2:1-20. [ Links ]
9. Paterson CR, Mole PA. Bone density in osteogenesis imperfecta may well be normal. Postgrad Med J 1994;70:104-7. [ Links ]
10. Gil Fournier, B. Importancia del diagnóstico genético en la Osteogénesis Imperfecta. Voces de cristal 2009:30-2. [ Links ]
11. Shapiro JR, Thompson CB, Wu Y, Nunes M, Gillen C. Bone Mineral Density and Fracture Rate in Response to Intravenous and Oral Bisphosphonates in Adult Osteogenesis Imperfecta. Calc Tissue Int 2010;87:120-9. [ Links ]
12. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2014;7:CD005088. [ Links ]
13. Hoyer-Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years' experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis 2014;9:145. [ Links ]
14. Orwoll ES, Shapiro J, Veith S, Wang Y, Lapidus J, Vanek C, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest 2014;124:491-8. [ Links ]
