










 Custom services
Custom services
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La osteoporosis y sus fracturas asociadas son el problema óseo postmenopáusico más común, y afecta a mujeres y hombres de todas las etnias. Los bisfosfonatos nitrogenados (N-BPs), incluyendo alendronato, risendronato, ibandronato y zolendronato, son el tratamiento más utilizado para la osteoporosis en millones de pacientes en todo el mundo. A pesar de la importante eficacia anti-fractura de los BPs, ampliamente demostrada en varios ensayos clínicos1 y revisiones sistemáticas2, se han descrito algunos efectos adversos poco frecuentes potencialmente asociados a su uso prolongado, entre ellos las fracturas atípicas de fémur (FAFs)3. Estas fracturas son no-traumáticas y están caracterizadas por su localización subtrocantérica o en la diáfisis del fémur, y frecuentemente son bilaterales4.
Los mecanismos patogénicos de las FAFs no son del todo conocidos, y se ha especulado mucho sobre sus causas. Se ha propuesto que una supresión excesiva de la resorción ósea por parte de los N-BPs podría contribuir a desencadenar una FAF pero su fisiopatología es compleja y se cree que hay otros factores importantes involucrados. Algunos factores de riesgo propuestos son el grosor cortical y la geometría pélvica5. Además, se han descrito casos de FAF en pacientes afectados por otras enfermedades óseas monogénicas, como la hipofosfatasia6, la osteogenesis imperfecta7 o el síndrome de osteoporosis pseudoglioma8.
Dada la baja incidencia de las FAFs en la población general (5,9 casos por 100.000 personas/año), podemos hipotetizar que hay unas causas genéticas raras subyacentes que pueden incrementar la susceptibilidad a las FAFs, y que pueden ocurrir espontáneamente o desencadenarse después de la interacción con los BPs. Actualmente no hay pruebas genéticas o bioquímicas que puedan ayudar a identificar los pacientes con un elevado riesgo a sufrir una FAF. La identificación de los determinantes genéticos de las FAFs ayudaría a esclarecer los mecanismos etiológicos, al desarrollo de herramientas de diagnóstico y de evaluación del riesgo de sufrir una FAF, y a posibles estrategias terapéuticas.
Anteriormente, identificamos 3 hermanas diagnosticadas con FAF que fueron tratadas con BPs durante más de 5 años9. Esta observación nos sugirió que podría haber un trasfondo genético que predispusiera a las FAFs relacionadas al uso prolongado de BPs. En consecuencia, llevamos a cabo la secuenciación del exoma completo de las 3 hermanas y de otras 3 pacientes no relacionadas para identificar mutaciones potencialmente relacionadas con las FAFs en estas pacientes. Identificamos 37 variantes raras compartidas por las 3 hermanas, una de las cuales se estudió en detalle9. En el presente trabajo describimos el conjunto de variantes encontradas y su posible interacción.
Material y métodos
Pacientes
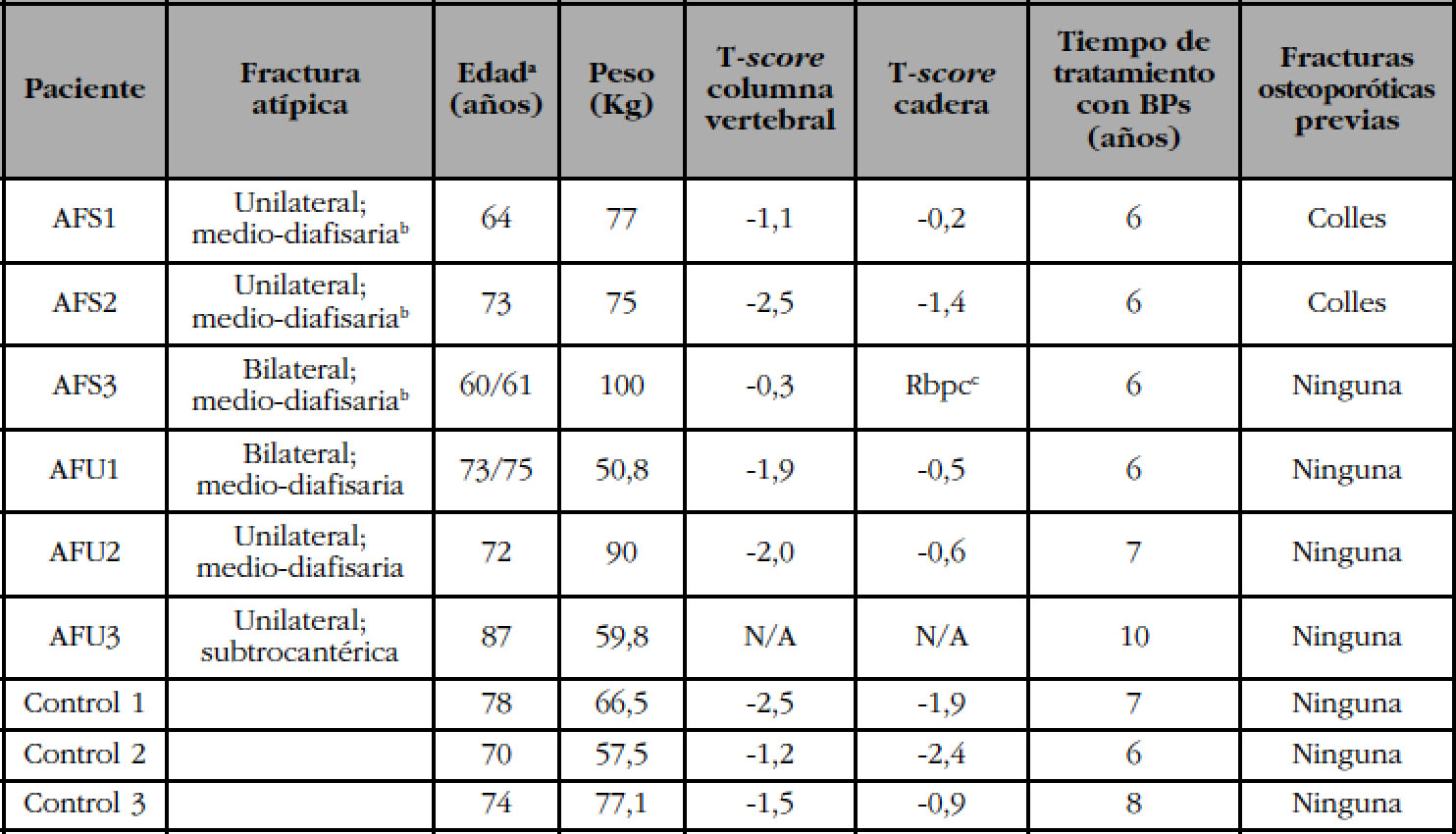
Se estudiaron seis pacientes con FAFs y que habían sido tratadas durante más de 5 años con BPs: 3 hermanas visitadas en el Hospital Universitario Reina Sofía (Córdoba, España) y 3 pacientes no relacionadas visitadas en el Hospital del Mar (Barcelona, España). Como controles, se estudiaron 3 pacientes tratadas con BPs por más de 6 años pero sin FAFs. Las características de pacientes y controles están descritas en la tabla 1. Las 3 hermanas afectas fueron tratadas con estatinas y recibían regularmente PPIs pero no habían sido tratadas con glucocorticoides ni ningún otro compuesto que afecte al hueso, aparte de los BPs. En el caso de las fracturas unilaterales, se realizaron pruebas radiológicas y RMN que descartaban la fractura contralateral. Se obtuvo consentimiento informado escrito de todas las pacientes, de acuerdo con la regulación del Comité Ético de Investigación Clínica del Parque de Salud Mar, que aprobó el estudio.
Secuenciación del exoma completo
Se extrajo ADN de sangre periférica de las pacientes con el kit Wizard Genomic DNA Purification (Promega) y se utilizó para secuenciar el exoma completo en el Centro Nacional de Análisis Genómico (CNAG) (Barcelona). Las librerías se generaron con el kit de captura de exones SureSelect XT Human All Exon; cat:5190-6208 (Agilent Technologies), después de haber fragmentado el ADN y ligado los adaptadores específicos de Agilent. La secuenciación paired-end (2x76 pb) se realizó en la plataforma Illumina HiSeq2000. Las imágenes del instrumento se procesaron utilizando el programa del fabricante para generar archivos de secuencia FASTQ.
El análisis bioinformático se llevó a cabo en la plataforma de Bioinformática para Enfermedades Raras (Bier) del CIBERER, en Valencia. Los archivos FASTQ se alinearon con el programa libre Burrows-Wheeler Aligner10 (http://bio-bwa.sourceforge.net/) utilizando el ensamblado del genoma humano de referencia GRCh37 (hg19)11. Las variantes de un solo nucleótido y los indels se identificaron utilizando el programa GATK12. Finalmente, para añadir a las variantes información sobre la frecuencia del alelo minoritario (minor allele frequency; MAF) proveniente de dbSNP y del proyecto 1000 Genomas (http://www.1000genomes.org)13, se utilizó la herramienta de anotación VARIANT14. Los datos se convirtieron al formato BAM (binary equivalent SAM) y se visualizaron mediante el programa Integrative Genomics Viewer (IGV) (http://www. broadinstitute.org/igv).
Las variantes genéticas se filtraron según las siguientes premisas: a) variante no-sinónima, b) no descrita previamente o con una MAF <0,005 en dbSNP y en el proyecto 1000 Genomas, c) no presente en NHLBI Go Exome Sequencing Project (ESP) (http://evs.gs.washington.eu/EVS/), y d) no presente en 8 exomas de individuos de la población general, obtenidos en nuestro laboratorio.
Inicialmente sólo se tuvieron en cuenta las mutaciones compartidas por las tres hermanas, tanto en un modelo de herencia dominante como recesivo. Después se priorizaron mutaciones en genes candidatos en las otras tres pacientes. Las puntuaciones de SIFT15, PolyPhen16 y de conservación evolutiva obtenidas de PhastCons17 se utilizaron para priorizar las variantes.
Validación de las variantes genéticas
Las mutaciones encontradas se validaron mediante PCR y secuenciación Sanger, que fue llevada a cabo bidireccionalmente utilizando el kit BigDyeTM v3.1 Terminator Cycle Sequencing (Applied Biosystems), según las instrucciones del fabricante. Los cebadores utilizados para la validación se diseñaron utilizando el programa OligoEvaluator (Sigma-Aldrich). Finalmente, las mutaciones validadas se buscaron en el Exome Aggregation Consortium (ExAC) para obtener sus frecuencias poblacionales, y se analizaron mediante secuenciación Sanger en las 3 mujeres controles.
Análisis in silico
Las mutaciones se localizaron en su contexto genético utilizando el UCSC Genome Browser (https:// genome.ucsc.edu/) y el Ensembl Genome Browser (http://www.ensembl.org/) y se extrajo información de los genes de GeneCards (http://www.gencards. org/) y BioGPS (http://biogps.org/). Se realizó un análisis de enriquecimiento funcional utilizando la herramienta bioinformática DAVID18 (https://david. ncifcrf.gov/).
El estudio funcional in silico de las proteínas mutadas se realizó utilizando Uniprot (http://uniprot.org), RCSB Protein Data Bank (PDB) (http:// www.rcsb.org/pdb) y Pfam (http://pfam.xfam.org). Los alineamientos de proteínas se realizaron utilizando el UCSC Genome Browser y los programas Clustal Omega (http://www.clustal.org/omega) y ESPript (http://espript.ibcp.fr).
Construcción de la red
La red de interacción de los genes FAF (AFFGeNet) se construyó según Boloc et al.19 para identificar genes o proteínas que interaccionan con los 37 genes FAF, considerados como genes driver (Tablas 2a y 2b), teniendo en cuenta las interacciones binarias y direccionales. Los datos de interacción high-throughput se obtuvieron de BioGRID (versión 3.4.133)20 y STRING [Search Tool for the Retrieval of Interacting Genes/Proteins] version 1021 y la red se enriqueció con información adicional de GeneOntology (http://geneontology. org), GeneCards, OMIM, UniProt, RefSeq, y UCSC.

Tabla 2b Otras variantes encontradas en las pacientes no relacionadas
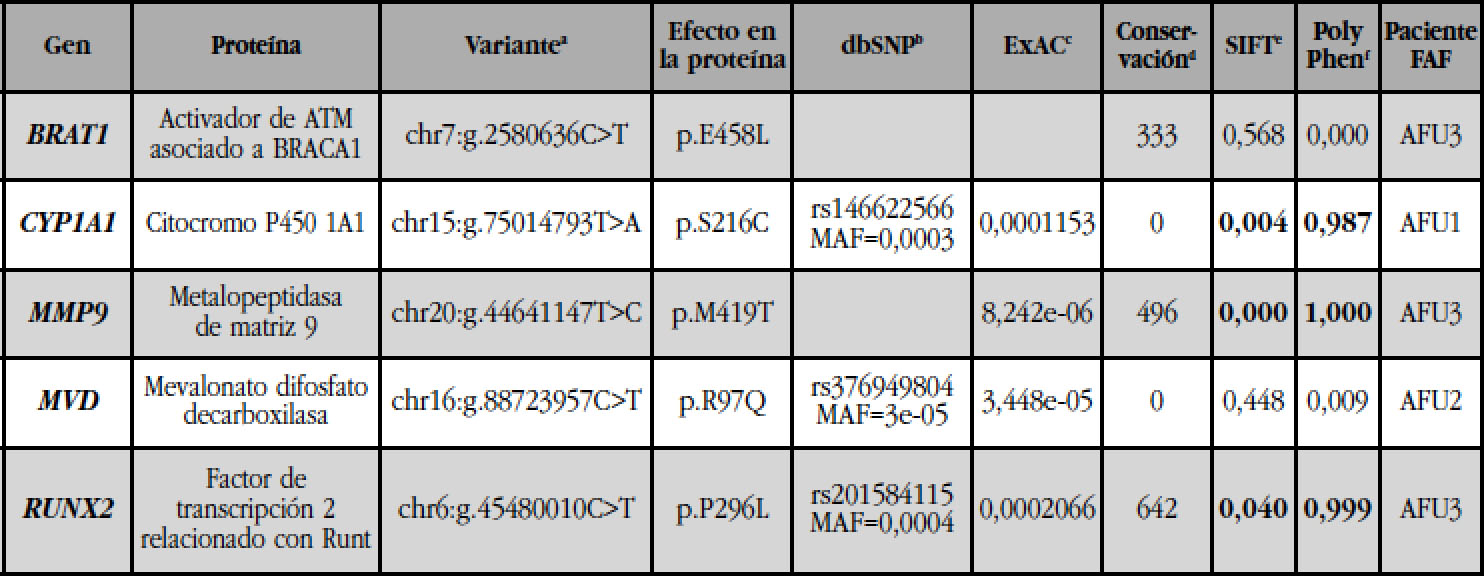
(a): posición genómica de la variante en el genoma de referencia humano GRCh37; (b): número de identificador de referencia del SNP (rs) y MAF (frecuencia del alelo minoritario) de las variantes descritas; (c): frecuencia alélica de las variantes descritas en la base de datos ExAC; (d): puntuación de conservación del PhastCons (0 a 1.000), siendo 1.000 el locus más conservado y 0 un locus no conservado; (e): SIFT: 0-0,05 perjudicial (en negrita); 0,051-1 tolerable; (f): PolyPhen: 0-0,4 benigno; 0,41-0,89 possiblemente perjudicial; 0,9-1 patogénico (en negrita); (*): presente en un alelo doble mutante.
Se implementó un script de Perl para capturar la sub-red de interacción utilizando los genes FAF para encontrar todos los caminos más cortos entre dos genes aplicando el algoritmo Dijkstra. La conectividad por parejas se analizó utilizando Circos22. El script produjo un gráfico esqueleto en formato JSON para poder visualizar los datos en la interfície web AFFGeNet (https://compgen.bio.ub. edu/AFFgenes, disponible bajo demanda). El formulario web contiene una entrada que se centra en los genes seleccionados, y la visualización de la red permite añadir o quitar nodos y mostrar información de los genes FAF. El color del borde identifica los nodos como drivers (lila), parejas upstream (verde) o downstream (azul) de los drivers seleccionados, y otros (gris). El color del interior de los nodos representa la expresión génica específica del hueso, que se obtuvo del Gene Expression Omnibus (GEO)23, concretamente de un estudio sobre células precursoras de osteoclastos tratadas o no tratadas con BPs (alendronato o risendronato) durante su diferenciación a osteoclasto maduro24 (GSE63009). La escala de colores va de amarillo intenso (subexpresado) a azul oscuro (sobreexpresado), siendo el blanco indicativo de ningún cambio de expresión.
Resultados
Variantes detectadas en la secuenciación del exoma completo en las 3 hermanas
Las tres hermanas (AFS1, AFS2, AFS3) y las 3 pacientes no relacionadas (AFU1, AFU2, AFU3) se analizaron separadamente.
Los exomas de las 3 hermanas se interseccionaron y no se identificó ninguna variante en homocigosis en común. Por el contrario, se identificaron 74 variantes en heterocigosis compartidas (coherentes con un modelo de herencia dominante), 37 de las cuales se validaron por secuenciación Sanger. En 3 de los genes (FN1, BRAT1 y XAB2), se encontraron 2 mutaciones diferentes. En los tres casos se pudo determinar que las variantes se encontraban en fase, siendo alelos doble-mutantes y no heterocigotos compuestos, mediante la visualización de los reads con el programa IGV y el análisis de polimorfismos intragénicos. Las 37 variantes compartidas por las 3 hermanas, todas ellas codificantes, se muestran en la tabla 2a, ordenadas según su puntuación de conservación. Se trata de variantes de cambio de sentido (n=35), una variante truncante y una deleción en fase. La primera variante de la lista, con la mejor puntuación de conservación y predicha como deletérea, se encuentra en el gen GGPS1, tal y como describimos anteriormente9.
Análisis de los genes mutados en las 3 pacientes no relacionadas
Los genes con variantes compartidas por las 3 hermanas (Tabla 2a) se analizaron en los exomas de las pacientes no relacionadas utilizando el programa IGV. Ninguna de las variantes de la Tabla 2a se encontró en las pacientes no relacionadas. No obstante, se encontraron otras dos variantes en los genes BRAT1 y CYP1A1, en las pacientes AFU3 y AFU1, respectivamente (Tabla 2b).
La variante de CYP1A1 presente en la paciente AFU1 (p.Ser216Cys) supone el cambio de una serina a una cisteína, en una posición cercana al sitio de unión al sustrato. Los predictores de patogenicidad sugirieron que este cambio es muy deletéreo para la función de la proteína. Igualmente, la variante de CYP1A1 presente en las tres hermanas (p.Arg98Trp) supone el cambio de un aminoácido básico (arginina) a un aminoácido aromático hidrofóbico (triptófano), en un giro de la proteína con puentes de hidrógeno. Por el contrario, las tres variantes encontradas en el gen BRAT1 (dos en las tres hermanas, en un alelo doble mutante, y una en la paciente AFU3) no afectan a la función de la proteína, según los predictores.
Análisis de genes candidatos en 3 pacientes no relacionadas
A continuación, se utilizó el programa IGV para analizar, en los exomas de las tres pacientes no relacionadas, distintos genes involucrados en el metabolismo óseo, la función osteoclástica y la vía del mevalonato. Se encontraron variantes en los genes MMP9 (AFU3), MVD (AFU2) y RUNX2 (AFU3), que se validaron por secuenciación Sanger (Tabla 2b). La mutación en el gen MMP9, que codifica la colagenasa de tipo IV, implica el cambio de una metionina (un aminoácido hidrofóbico con un grupo que contiene azufre) a una treonina (aminoácido hidrofílico) en la posición 419, dentro del dominio catalítico. Esta variante aparece en la base de datos ExAC, con una frecuencia alélica muy baja (8,2e-06), y perjudicial para la función de la proteína, según SIFT y PolyPhen predijeron que probablemente per-SIFT y PolyPhen. La mutación en RUNX2 es una judica su función. El gen MVD codifica la enzima substitución de una prolina, un aminoácido cíclico, mevalonato 5-difosfato decarboxilasa, de la vía del por una leucina, un aminoácido alifático hidrofóbimevalonato. La variante encontrada (p.Arg97Gln; co, en la posición 296, dentro de una región rica en rs376949804) supone el cambio de un aminoácido prolinas, serinas y treoninas. Este cambio, descrito básico a un aminoácido neutro y está presente en la en dbSNP (rs20184115), tiene una MAF=0,0004 y base de datos ExAC, también con una frecuencia probablemente afecta la función de la proteína, alélica muy baja (3,4e-05). Se trata de un cambio no según los predictores.
Análisis de las variantes en individuos controles y en la población general
Ninguna variante de las tablas 2a y 2b fue encontrada en 3 controles (pacientes tratadas con BPs durante un período largo de tiempo pero sin FAFs). Todas las variantes detectadas en las pacientes con FAF se buscaron en la base de datos ExAC para determinar si se trataba de variantes nuevas o muy raras (MAF <0,005). En ese sentido, once mutaciones no se encontraron ni en dbSNP ni en ExAC (GGPS1: p.D188Y; COG4: p.G85D; PGRMC1: p.P177H; TMEM25: p.V239del; HEPHL1: p.W991*; CUL9: p.T423I; IQCF6: p.R61W; MGA: p.S571L; SHC4: p.H180N; SMS: p.G14R; BRAT1: p.E458L). Las otras variantes tienen frecuencias ≤1/10000, según ExAC.
Red de interacción génica/proteica y enriquecimiento de vías
Se construyó una red de interacciones entre genes y/o proteínas para investigar las vías funcionales relacionadas con los 37 genes mutados encontrados en la secuenciación de los exomas y detectar otros genes potencialmente causales, así como mecanismos moleculares que puedan estar implicados en la generación de las FAFs. La figura 1 muestra la conectividad entre parejas de genes. En distintos círculos, se muestran las conexiones de entrada y de salida para los 37 genes a distancias 1 a 4, respectivamente. A distancia 1 casi no hay interacciones, siendo FN1 el único gen conectado con otros. A distancia 2 se observa más conectividad. La mayoría de la conectividad entre parejas de genes se observa a distancia 3. El único gen que no presenta ninguna interacción a ningún nivel es IQCF6.

Figura 1 Esquema de la conectividad entre parejas de genes a distancias 1 a 4. En los círculos se muestran los símbolos de los 37 genes FAF encontrados en este estudio y sus conexiones de entrada y de salida
La red de interacciones de genes/proteínas muestra que GGPS1 y CYP1A1, dos de los genes driver más relevantes, se conectan a distancia 3, a través de INS y IL6 (Figura 2a). Otros 4 genes driver (RUNX2, MVD, MMP9 y PGRMC1) están conectados con GGPS1 a distancia 2. MMP9 también está a distancia 2 de CYP1A1. Además, FN1 y MMP9 están conectados a distancia 1. De manera similar, los genes driver SYDE2 y NGEF están interconectados a distancia 2, a través de RHOB (Figura 2b).
El análisis de enriquecimiento de vías en los 37 genes mutados, realizado con la herramienta DAVID, dio como resultado la vía de biosíntesis de los isoprenoides (GO:0008299) (p=0,0006), que contiene los genes GGPS1, MVD y CYP1A1.
Discusión
En este trabajo hemos estudiado el trasfondo genético de 3 hermanas con FAF y 3 pacientes adicionales, no relacionadas, a través de la secuenciación masiva del exoma para identificar posibles genes de susceptibilidad a la patología. Hemos identificado 37 variantes raras (en 34 genes) compartidas por las 3 hermanas, algunas de ellas no descritas anteriormente y consideradas dañinas por los predictores. La variante más llamativa fue la mutación p.Asp188Tyr en el gen GGPS1, que presentó la mejor puntuación de conservación, y que ya hemos descrito en un trabajo previo9. Otro hallazgo interesante fueron las dos mutaciones en el gen CYP1A1, una encontrada en las tres hermanas y la otra en una paciente no relacionada. Sin embargo, hay otras variantes que también podrían estar involucradas, en distintos grados, en la susceptibilidad a las FAFs asociadas a BPs o en el fenotipo osteoporótico subyacente, de modo que nuestros datos serían compatibles con un modelo en el cual la acumulación de variantes de susceptibilidad podría contribuir a la base genética de las FAFs.
Los estudios epidemiológicos sugieren que existe una relación entre las FAFs y un tratamiento prolongado con BPs. Shane et al., describieron períodos de tratamiento de una mediana de 7 años4. El riesgo absoluto de sufrir una FAF asociada al tratamiento con BPs se encuentra entre 2 casos por
100.000 pacientes/año a los 2 años de tratamiento y 78 casos por 100.000 pacientes/año a los 8 años de tratamiento25. Estos datos sugieren que la duración de la terapia con BPs influiría positivamente en el riesgo de sufrir estas fracturas. En nuestro estudio, los casos de 6 pacientes con FAF después de un tratamiento a largo plazo con BPs son consistentes con esta asociación. Además, la ocurrencia de las FAFs en las 3 hermanas sugiere una predisposición genética con un papel determinante en la patología. Este estudio ha sido el primer análisis de exoma de pacientes de FAF. Hemos priorizado mutaciones raras, no-sinónimas, compartidas por las 3 hermanas. No se encontró ninguna mutación en homocigosis o heterocigosis compuesta en ningún gen. Estos hallazgos van en contra de un patrón de herencia recesivo para estos casos y son consistentes con el hecho que la FAF no es una enfermedad genética severa que ocurra durante las primeras etapas de la vida. No obstante, en el modelo dominante, se encontraron 34 genes mutados, algunos muy importantes para el metabolismo óseo. En un trabajo anterior que tenía por objetivo descubrir las causas genéticas de las FAFs, se utilizó un chip de exoma con >300.000 variantes codificantes ya conocidas y se encontraron 21 variantes raras sobrerrepresentadas en 13 pacientes de FAF26. Sin embargo, ninguno de estos alelos de riesgo se encontró en los pacientes analizados en nuestro estudio. En concreto, no se encontraron variantes en el gen PPEF2, el único con un cambio asociado significativamente con el fenotipo en el estudio de Pérez-Núñez et al.26 Esto apunta a una base genética heterogénea para las FAFs. En todo caso, es importante señalar que nuestra aproximación metodológica difiere de la del estudio mencionado en tanto que analizamos toda la secuencia del exoma, cosa que nos permitió encontrar variantes no descritas anteriormente.
En el presente estudio, el único gen con mutaciones en las 3 hermanas y en pacientes no relacionados fue CYP1A1. Recientemente, Peris et al.27 secuenciaron este gen en 17 pacientes de FAF y encontraron otra mutación en una de ellas. El gen CYP1A1 codifica la enzima citocromo P450 1A1 que está involucrada en el metabolismo de fármacos y xenobióticos. Se trata de una hidroxilasa de hidrocarburos arilos y sus sustratos exógenos potenciales incluyen hidrocarburos aromáticos policíclicos, y está implicada en la formación de distintos tipos de cáncer humanos. Sus sustratos endógenos incluyen eicosanoides, que pueden generar productos biológicamente activos que actúan en el sistema vascular, entre otros. Este gen también es responsable de la hidroxilación del 17β-estradiol, la estrona y la vitamina D en tejidos extrahepáticos28. Esto es coherente con su papel en la biología ósea, una idea apoyada por Napoli et al.29, quienes demostraron que el polimorfismo C4887A estaba relacionado con un aumento significativo del catabolismo de los estrógenos y con una densidad mineral ósea (DMO) femoral baja en mujeres postmenopáusicas. Por lo tanto, CYP1A1 se presenta como otro gen de susceptibilidad potencial a las FAFs, aunque el mecanismo exacto de su acción en el metabolismo óseo todavía es desconocido y más estudios son necesarios para elucidarlo.
Entre los otros genes con variantes en las tres hermanas, FN1 codifica la fibronectina, una proteína de la matriz extracelular necesaria para la regulación de la deposición del colágeno de tipo I por parte de los osteoblastos, esencial para la mineralización de la matriz extracelular, y cuyos niveles se han visto afectados por el tratamiento con BPs30. Encontramos que las tres hermanas eran portadoras de un alelo doble mutante (p.V2241I y p.R1496W) en FN1, donde las dos mutaciones fueron consideradas como dañinas por los predictores de patogenicidad. Esta fibronectina alterada podría afectar la mineralización ósea y/o la respuesta a los BPs y estar relacionada con el riesgo a sufrir una FAF en estas mujeres. También encontramos mutados 2 reguladores de GTPasas pequeñas: SYDE2 y NGEF. Sus funciones respectivas (activación de las GTPasas RHO y de intercambio de sus nucleótidos de guanina) constituyen pistas sobre posibles efectos en la función osteoclástica y en la respuesta a los BPs. Las RHO GTPasas están en la vía del mevalonato en una posición por debajo del sitio de acción de los BPs, ya que tienen que ser preniladas (farnesiladas o geranilgeraniladas) para su correcta función celular. Por otra parte, nuestra red de interacción de genes/proteínas muestra como NGEF está muy relacionado con las efrinas y los receptores de efrinas (Figura 2b), que tienen un papel clave en el mecanismo de acoplamiento entre osteoclastos y osteoblastos31. Otro grupo de genes mutados en las 3 hermanas codifican proteínas nucleares con efectos pleiotrópicos sobre la expresión génica y/o la reparación del DNA (KDM4C, XAB2, NVL, NKAP, ERCC6L2). De ellos destacamos el gen KDM4C, que codifica una demetilasa lisina-específica que contiene un dominio JmjC, que ha sido previamente asociado con la edad de menarquia32, un biomarcador para la densidad ósea.

Figura 2 Detalles de la red de interacción entre genes/pro-estas pacientes en dos proteínas claves teínas. El color del interior de los nodos indica la subexpre-para el remodelado óseo (RUNX2 y MMP9) sión (amarillo), sobreexpresión (azul) o ningún cambio de expresión (blanco) en osteoclastos tratados con alendrona to o risendronato (datos de Yuen et al., 201424). El color externo identifica los genes como drivers (mutados en nuestras pacientes) en lila, upstream de los genes mutados en verde, y otros en gris. a) Interacciones de los genes GGPS1 y CYP1A1 a distancia 2 (y algunas del gen MMP9 a distancia 1). Nota: algunas conexiones se han omitido para la cla-matriz extracelular ósea37, afectando a la ridad de la figura. En particular, los nodos RUNX2 y FN1 no arquitectura del hueso trabecular y a la se han expandido para mostrar todos sus conectores. b) estructura del hueso cortical38. Por estas Interacciones de los genes SYDE2 y NGEF a distancia 1
Otros genes encontrados mutados en las hermanas fueron el gen PGRMC1 que codifica el componente 1 del receptor de membrana de la progesterona, y que fue previamente asociado al fallo ovárico prematuro33; el gen COG4 (que codifica la subunidad 4 del complejo oligomérico conservado del Golgi), relevante dada la importancia del transporte de vesículas a través del Golgi en los osteoclastos34; y el gen EML1(que codifica una proteína asociada a microtúbulos) que puede ser importante en relación al cilio primario en osteocitos35. En conjunto, las funciones y conocimiento previo de 13 de los 34 genes mutados en las 3 hermanas concuerdan con su posible implicación en la patología. Estas mutaciones se buscaron en las 3 pacientes de FAF no relacionadas, con resultados negativos.
No obstante, mediante una aproximación de genes candidatos, se encontraron mutaciones en estas pacientes en dos proteínas claves para el remodelado óseo (RUNX2 y MMP9) y en otra enzima de la vía del mevalonato (MVD, mevalonato difosfato carboxilasa). RUNX2 es un factor de transcripción esencial para la diferenciación osteoblástica36, mientras que MMP9 es una metaloproteasa expresada en osteoclastos que degrada la matriz extracelular ósea37, afectando a la arquitectura del hueso trabecular y a la estructura del hueso cortical38. Por estas razones, ambos pueden estar involucrados en el riesgo a la FAF. Es sabido que RUNX2 activa la expresión génica de MMP939 y esta interacción puede tener efectos sinérgicos en las propiedades biomecánicas del hueso en la paciente AFU3, que tiene ambas mutaciones (Nota: esta interacción no se muestra en la Figura 2a para que otras interacciones se puedan mostrar claramente). Finalmente, en la paciente AFU2 se encontró una mutación de cambio de sentido en el gen MVD, añadiendo una segunda proteína mutada de la vía del mevalonato. En la figura 3 se muestran, en el contexto de las células óseas, las proteínas codificadas por los genes que hemos encontrado mutados y cuya función en el hueso se conoce o está predicha.

Figura 3 Proteínas codificadas por los genes mutados en las pacientes de FAF de este estudio y relacionadas con la función ósea
En conjunto, todas estas variantes raras pueden formar parte de un trasfondo genético asociado al desarrollo de los cambios óseos que dan lugar a las FAFs y a la posible interacción negativa con los BPs. Es probable que varios genes con efectos aditivos pequeños, y sus interacciones, estén implicados en las FAFs relacionadas con los BPs. Además, cada paciente individual podría ser portador de distintas variantes genéticas específicas.
Los puntos fuertes de este estudio son la posibilidad de analizar 3 hermanas con FAF y el abordaje por secuenciación del exoma completo, que carece de hipótesis previa. En este sentido, pudimos identificar mutaciones dañinas en genes que pertenecen a la vía del mevalonato, así como otros genes relacionados con el metabolismo óseo. Por otro lado, el bajo número de pacientes y controles estudiados es una limitación del estudio y serán necesarios más estudios de secuenciación del exoma de pacientes de FAF adicionales y de pacientes no fracturados con un tratamiento a largo plazo con BPs (actuando como controles) para clarificar el papel preciso de estos genes y mutaciones. A pesar de la plausibilidad biológica del efecto dañino de las mutaciones encontradas, se necesita la replicación de estos hallazgos.
La identificación del trasfondo genético para las fracturas atípicas de fémur abre la puerta al futuro desarrollo de herramientas de diagnóstico y predicción del riesgo a sufrir este tipo de fracturas para determinar la idoneidad del tratamiento con BPs.

