










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.10 no.4 Madrid nov./dic. 2018 Epub 03-Abr-2023
https://dx.doi.org/10.4321/s1889-836x2018000400002
Original Articles
Genetic study of atypical femoral fractures using exome sequencing in three affected sisters and three unrelated patients
1Departamento de Genética, Microbiología y Estadística - Facultad de Biología - Universidad de Barcelona - Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) - Instituto de Salud Carlos III (ISCIII) - Instituto de Biomedicina de la Universidad de Barcelona (IBUB) - Instituto de Investigación Sant Joan de Déu (IRSJD) - Barcelona (España)
2Unidad de Investigación en Fisiopatología Ósea y Articular (URFOA); Instituto Hospital del Mar de Investigaciones Médicas (IMIM) - Parque de Salud Mar - Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES); Instituto de Salud Carlos III (ISCIII) - Barcelona (España)
3Departamento de Genética, Microbiología y Estadística; Facultad de Biología; Universidad de Barcelona - Instituto de Biomedicina de la Universidad de Barcelona (IBUB) -Barcelona (España)
4Unidad de Metabolismo Mineral; Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC); Hospital Universitario Reina Sofía - Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES); Instituto de Salud Carlos III (ISCIII) - Córdoba (España)
5Grupo de Investigación en Enfermedades Prevalentes del Aparato Locomotor (GREMPAL) - Instituto de Investigación en Atención Primaria (IDIAP) Jordi Gol - Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES) - Universidad Autónoma de Barcelona (UAB) - Barcelona (España)
6Instituto Nacional para la Investigación de la Salud (NIHR); Unidad de Investigación Biomédica (BRU) Musculoesquelética y Centro de Investigación Botnar; Departmento de Ortopedia, Reumatología y Ciencias Músculoesqueléticas Nuffield - Universidad de Oxford - Oxford (Reino Unido)
Objectives:
Atypical femoral fractures (AFF) are rare, often related to long-term bisphosphonate (BPs) treatment. Their pathogenic mechanisms are not precisely known and there is no evidence to identify patients with a high risk of AFF. The aim of this work is to study the genetic bases of AFFs.
Materials and methods:
Whole-exome sequencing was carried out on 3 sisters and 3 unrelated additional patients, all treated with BPs for more than 5 years. Low frequency, potentially pathogenic variants shared by the 3 sisters, were selected, were selected and a network of gene and protein interactions was constructed with the data found.
Results:
We identified 37 rare variants (in 34 genes) shared by the 3 sisters, some not previously described. The most striking variant was the p.Asp188Tyr mutation in the enzyme geranylgeranyl pyrophosphate synthase (encoded by the GGPS1 gene), from the mevalonate pathway and essential for osteoclast function. Another noteworthy finding was two mutations (one in the 3 sisters and one in an unrelated patient) in the CYP1A1 gene, involved in the metabolism of steroids. We identified other variants that could also be involved in the susceptibility to AFFs or in the underlying osteoporotic phenotype, such as those present in the SYDE2, NGEF, COG4 and FN1 genes.
Conclusions:
Our data are compatible with a model where the accumulation of susceptibility variants could participate in the genetic basis of AFFs.
Key words atypical femoral fractures; bisphosphonates; GGPS1; CYP1A1; whole-exome sequencing
Introduction
Osteoporosis and its associated fractures are the most common postmenopausal bone problems, affecting women and men of all ethnic groups. Nitrogen-containing bisphosphonates (N-BPs), including alendronate, risendronate, ibandronate and zolendronate figure as the most widely used osteoporosis treatments in millions of patients worldwide. Despite the significant anti-fracture efficacy of BPs, which has been widely demonstrated in several clinical trials1 and systematic reviews2, some infrequent adverse effects associated with prolonged use have been described, including atypical femur fractures (AFFs)3. These fractures are non-traumatic and characterized by their subtrochanteric location or in the diaphysis of the femur, and are frequently bilateral4.
AFFs’ pathogenic mechanisms are not completely known and much has been speculated about their causes. An excessive suppression of bone resorption by N-BPs could trigger an AFF but its pathophysiology is complex and other important factors are reportedly involved. Some proposed risk factors are cortical thickness and pelvic geometry5. In addition, cases of AFF have been described in patients affected by other monogenic bone diseases, such as hypophosphatasia6, osteogenesis imperfecta7 or the syndrome of osteoporosis pseudoglioma8.
Given the low incidence of AFFs in the general population (5.9 cases per 100,000 people/year), we can hypothesize that there are underlying rare genetic causes that may increase susceptibility to AFFs, and that they might occur spontaneously or be triggered after the interaction with the BPs. Currently, there are no genetic or biochemical tests to help identify patients with a high risk of AFF. Identifying the genetic determinants of AFFs would help to clarify the etiological mechanisms, develop diagnostic tools and evaluate AFF risk and possible therapeutic strategies.
Previously, we identified 3 sisters diagnosed with AFF who were treated with BPs for more than 5 years9. This observation suggested that there might be a genetic background predisposing to AFFs related to prolonged use of BPs. Consequently, we carried out the sequencing of the complete exome of the 3 sisters and of 3 other unrelated patients to identify mutations potentially related to the AFFs in these patients. We identified 37 rare variants shared by the 3 sisters, one of which was studied in detail9. In the present work, we describe the set of variants found and their possible interaction.
Material and methods
Patients
Six patients with AFFs who had been treated for more than 5 years with BPs were studied: 3 sisters visited at the Reina Sofía University Hospital (Córdoba, Spain) and 3 unrelated patients visited at the Hospital del Mar (Barcelona, Spain). As controls, 3 patients treated with BPs for more than 6 years but without AFFs were studied. The characteristics of patients and controls are described in Table 1. The 3 affected sisters were treated with statins and received PPIs regularly but had not been treated with glucocorticoids or any other compound that affects the bone, apart from the BPs. In the case of unilateral fractures, radiological and MRI tests were performed that ruled out the contralateral fracture. Written informed consent was obtained from all patients, in accordance with the regulations of the Clinical Research Ethics Committee of the Mar Health Park, which approved the study.

Complete exome sequencing
Peripheral blood DNA was extracted from the patients with the Wizard Genomic DNA Purification kit (Promega) and used to sequence the complete exome at the National Center for Genomic Analysis (CNAG) (Barcelona). Libraries were generated with the SureSelect XT Human All Exon exons capture kit; cat: 5190-6208 (Agilent Technologies), after having fragmented the DNA and ligated the specific Agilent adapters. Pairedend sequencing (2x76 bp) was carried out on the Illumina HiSeq2000 platform. The images were processed using the manufacturer's program to generate FASTQ sequence files.
The bioinformatic analysis was carried out in the Bioinformatics platform for Rare Diseases (Bier) of the CIBERER, in Valencia. The FASTQ files were aligned with the free program Burrows-Wheeler Aligner10 (http://bio-bwa.sourceforge.net/) using the reference human genome assembly GRCh37 (hg19)11. Single-nucleotide and indel variants were identified using the GATK program12. Finally, to add to the variants information on the frequency of the minority allele (MAF) from dbSNP and the 1000 Genomes project (http://www.1000genomes.org)13, the annotation tool VARIANT was used14. The data were converted to the BAM (binary equivalent SAM) format and visualized using the Integrative Genomics Viewer (IGV) program (http://www. Broadinstitute.org/igv).
The genetic variants were filtered according to the following premises: a) non-synonymous variant, b) not previously described or with an MAF <0.005 in dbSNP and in the 1000 Genomes project, c) not present in the NHLBI Go Exome Sequencing Project (ESP) (http://evs.gs.washington.eu/EVS/), and d) not present in 8 exomes of individuals from the general population, obtained in our laboratory.
Initially, only the mutations shared by the three sisters were considered, both in a model of dominant and recessive inheritance. Next, mutations in candidate genes were prioritized in the other three patients. The SIFT15, PolyPhen16 and evolutionary conservation scores obtained from PhastCons17 were used to prioritize the variants.
Validation of genetic variants
The mutations found were validated by PCR and Sanger sequencing, which was carried out bidirectionally using the BigDyeTM v3.1 Terminator Cycle Sequencing kit (Applied Biosystems), according to the manufacturer's instructions. The primers used for the validation were designed using the OligoEvaluator program (Sigma-Aldrich). Finally, the validated mutations were searched in the Exome Aggregation Consortium (ExAC) to obtain their population frequencies, and analyzed by Sanger sequencing in the 3 control women.
In silico analysis
Mutations were localized in their genetic context using the UCSC Genome Browser (https://genome.ucsc.edu/) and the Ensembl Genome Browser (http://www.ensembl.org/) and extracted information from GeneCards genes (http://www.gencards.org/) and BioGPS (http://biogps.org/). A functional enrichment analysis was carried out using the bioinformatics tool DAVID18 (https: //david. Ncifcrf.gov/).
In silico functional study of the mutated proteins was carried out using Uniprot (http://uniprot.org), RCSB Protein Data Bank (PDB) (http:// www.rcsb.org/pdb) and Pfam (http:// pfam.xfam.org). The protein alignments were made using the UCSC Genome Browser and the Clustal Omega programs (http://www.clustal.org/omega) and ESPript (http://espript.ibcp.fr).
Construction of the network
The AFF gene interaction network (AFFGeNet) was constructed according to Boloc et al.19 to identify genes or proteins that interact with the 37 FAF genes, considered as driver genes (Tables 2a and 2b), taking into account the binary and directional interactions. The high-throughput interaction data were obtained from BioGRID (version 3.4.133)20 and STRING [Search Tool for the Retrieval of Interacting Genes/Proteins] version 1021 and the network was enriched with additional information from GeneOntology (http://geneontology.org), GeneCards, OMIM, UniProt, RefSeq, and UCSC.

table 2b Other variants found in unrelated patients
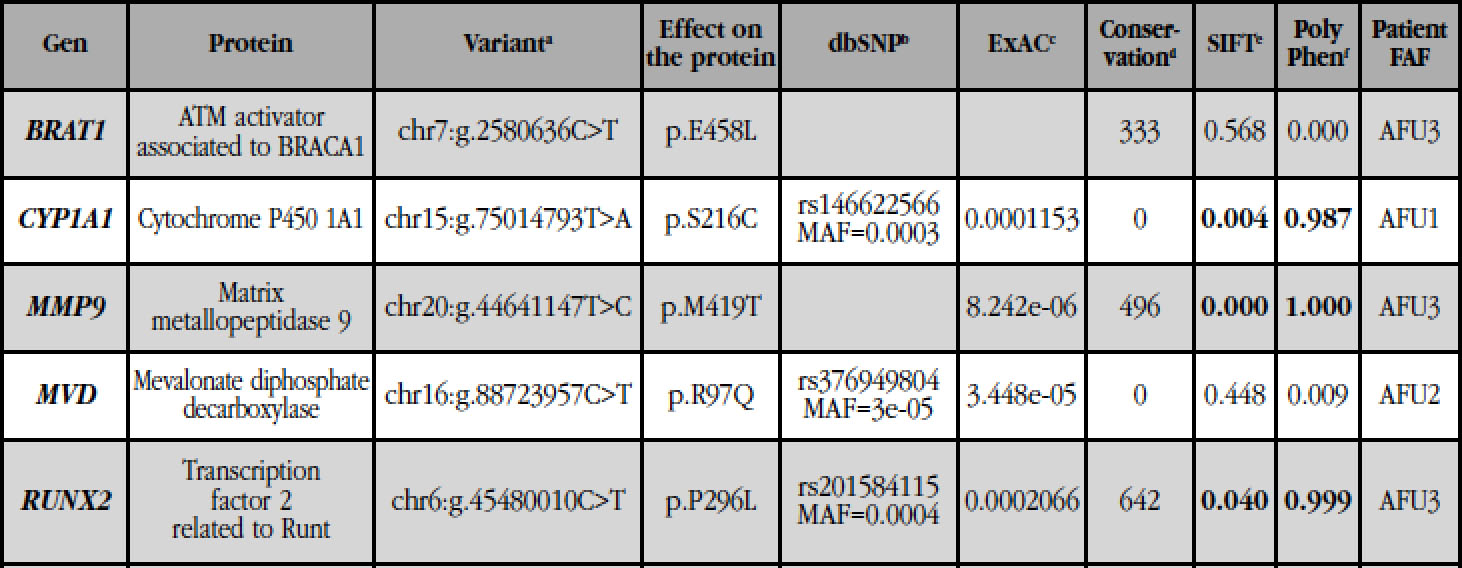
(a): genomic position of the variant in the human reference genome GRCh37; (b): reference identifier number of the SNP (rs) and MAF (minority allele frequency) of the variants described; (c): allelic frequency of the variants described in the ExAC database; (d): PhastCons conservation score (0 to 1,000), with 1,000 being the most conserved locus and 0 a non-conserved locus; (e): SIFT: 0-0.05 harmful (in bold); 0.051-1 tolerable; (f): PolyPhen: benign 0-0.4; 0.41-0.89, possibly harmful; 0.9-1 pathogenic (in bold); (*): present in a mutant double allele.
A Perl script was implemented to capture the interaction sub-network using the AFF genes to find all the shortest paths between two genes by applying the Dijkstra algorithm. The connectivity in pairs was analyzed using Circos22. The script produced a skeleton graphic in JSON format in order to visualize the data in the AFFGeNet web interface (https://compgen.bio.ub.edu/AFFgenes, available on demand). The web form contains an entry that focuses on the selected genes, and the visualization of the network allows you to add or remove nodes and display information of the AFF genes. The border color identifies the nodes as drivers (lilac), upstream (green) or downstream (blue) pairs of the selected drivers, and others (gray). The color of the interior of the nodes represents the specific gene expression of the bone, which was obtained from the Gene Expression Omnibus (GEO)23, specifically from a study on osteoclast precursor cells treated or not treated with BPs (alendronate or risendronate) during their differentiation to mature osteoclast24 (GSE63009). The color scale goes from intense yellow (underexpressed) to dark blue (overexpressed), being the target indicative of no change of expression.
Results
Variants detected in the sequencing of the complete exome in the 3 sisters
The three sisters (AFS1, AFS2, AFS3) and the 3 unrelated patients (AFU1, AFU2, AFU3) were analyzed separately.
The exomes of the 3 sisters intersected and no variant in common homozygosis was identified. On the contrary, 74 variants were identified in shared heterozygosis (consistent with a dominant inheritance model), 37 of which were validated by Sanger sequencing. In 3 of the genes (FN1, BRAT1 and XAB2), 2 different mutations were found. In all three cases it was possible to determine that the variants were in phase, being double-mutant alleles and non-heterozygous compounds, by visualizing the reads with the IGV program and the analysis of intragenic polymorphisms. The 37 variants shared by the 3 sisters, all of them coding, are shown in table 2a, ordered according to their conservation score. These are change-of-sense variants (n=35), a truncating variant and a phase deletion. The first variant of the list, with the best conservation score and predicted as deleterious, is found in the GGPS1 gene, as described above9.
Analysis of mutated genes in the 3 unrelated patients
The genes with variants shared by the 3 sisters (Table 2a) were analyzed in the exomes of the unrelated patients using the IGV program. None of the variants of Table 2a was found in unrelated patients. However, two other variants were found in the BRAT1 and CYP1A1 genes, in patients AFU3 and AFU1, respectively (Table 2b).
The variant of CYP1A1 present in the patient AFU1 (p.Ser216Cys) involves the change of a serine to a cysteine, in a position close to the site of binding to the substrate. Predictors of pathogenicity suggested that this change is very deleterious for protein function. Similarly, the variant CYP1A1 present in the three sisters (p.Arg98Trp) involves the change of a basic amino acid (arginine) to a hydrophobic aromatic amino acid (tryptophan), in a protein spin with hydrogen bonds. In contrast, the three variants found in the BRAT1 gene (two in the three sisters, in a mutant double allele, and one in the patient AFU3) do not affect the function of the protein, according to the predictors.
Analysis of candidate genes in 3 unrelated patients
Next, the IGV program was used to analyze, in the exomes of the three unrelated patients, different genes involved in bone metabolism, osteoclastic function and the mevalonate pathway. Variants were found in the MMP9 (AFU3), MVD (AFU2) and RUNX2 (AFU3) genes, which were validated by Sanger sequencing (Table 2b). The mutation in the MMP9 gene, which encodes type IV collagenase, involves the change of a methionine (a hydrophobic amino acid with a sulfur-containing group) to a threonine (hydrophilic amino acid) at position 419, within the catalytic domain. This variant appears in the ExAC database, with a very low allelic frequency (8.2e-06), and SIFT and PolyPhen predicted that it probably impairs its function. The MVD gene encodes the enzyme mevalonate 5-diphosphate decarboxylase, from the mevalonate pathway. The variant found (p.Arg97Gln; rs376949804) involves the change from a basic amino acid to a neutral amino acid and is present in the ExAC database, also with a very low allelic frequency (3,4e-05). It is a change not harmful to the function of the protein, according to SIFT and PolyPhen. The mutation in RUNX2 is a substitution of a proline, a cyclic amino acid, by a leucine, a hydrophobic aliphatic amino acid, at position 296, within a region rich in prolines, serines and threonines. This change, described in dbSNP (rs20184115), has an MAF=0.0004 and probably affects the function of the protein, according to the predictors.
Analysis of the variants in control individuals and in the general population
No variant of tables 2a and 2b was found in 3 controls (patients treated with BPs for a long period of time but without AFFs). All the variants detected in patients with AFF were searched in the ExAC database to determine if they were new or very rare variants (MAF <0.005). In this sense, eleven mutations were found neither in dbSNP nor in ExAC (GGPS1: p.D188Y, COG4: p.G85D, PGRMC1: p.P177H, TMEM25: p.V239del, HEPHL1: p.W991*; CUL9: p .T423I; IQCF6: p.R61W; MGA: p.S571L; SHC4: p.H180N; SMS: p.G14R; BRAT1: p.E458L). The other variants have frequencies ≤1/10000, according to ExAC.
Network of gene/protein interaction and path enrichment
A network of interactions between genes and/or proteins was constructed to investigate the functional pathways related to the 37 mutated genes found in the sequencing of the exomes and detect other potentially causative genes, as well as molecular mechanisms that may be involved in the generation of the genes. AFFs. Figure 1 shows the connectivity between gene pairs. In different circles, the input and output connections for the 37 genes are shown at distances 1 to 4, respectively. At distance 1 there are almost no interactions, with FN1 being the only gene connected to others. At distance 2 more connectivity is observed. The majority of the connectivity between pairs of genes is observed at a distance. The only gene that does not present any interaction at any level is IQCF6.

Figure 1 Diagram of connectivity between gene pairs at distances 1 to 4. In the circles the symbols of the 37 AFF genes found in this study and their input and output connections are shown
The network of gene/protein interactions shows that GGPS1 and CYP1A1, two of the most relevant driver genes, are connected at distance 3, through INS and IL6 (Figure 2a). Another 4 driver genes (RUNX2, MVD, MMP9 and PGRMC1) are connected to GGPS1 at distance 2. MMP9 is also remote 2 of CYP1A1. In addition, FN1 and MMP9 are connected remotely 1. Similarly, the driver genes SYDE2 and NGEF are interconnected at distance 2, via RHOB (Figure 2b).
The path enrichment analysis in the 37 mutated genes, carried out with the DAVID tool, resulted in the isoprenoid biosynthesis pathway (GO: 0008299) (p=0.0006), which contains the GGPS1. MVD and CYP1A1 genes.

Figure 2 Details of the interaction network between genes/proteins. The color of the interior of the nodes indicates subexpression (yellow), overexpression (blue) or no change of expression (white) in osteoclasts treated with alendronate or risendronate (data from Yuen et al., 201424). The external color identifies the genes as drivers (mutated in our patients) in lilac, upstream of the genes mutated in green, and others in gray. a) Interactions of the GGPS1 and CYP1A1 genes at distance 2 (and some of the MMP9 gene at distance 1). Note: some connections have been omitted for the clarity of the figure. In particular, nodes RUNX2 and FN1 have not been expanded to show all their connectors. b) Interactions of the SYDE2 and NGEF genes at a distance 1
Discussion
In this work, we have studied the genetic background of 3 sisters with AFF and 3 additional patients, unrelated, through the massive sequencing of the exome to identify possible susceptibility genes to the pathology. We have identified 37 rare variants (in 34 genes) shared by the 3 sisters, some of them not previously described and considered harmful by the predictors. The most striking variant was the mutation p.Asp188Tyr in the GGPS1 gene, which presented the best conservation score, and which we have already described in a previous work9. Another interesting finding was the two mutations in the CYP1A1 gene, one found in the three sisters and the other in an unrelated patient. However, there are other variants that could also be involved, to varying degrees, in the susceptibility to AFFs associated with BPs or in the underlying osteoporotic phenotype, so that our data would be compatible with a model in which the accumulation of variants of susceptibility could contribute to the genetic base of the AFFs.
Epidemiological studies suggest that AFFs are related to prolonged treatment with BPs. Shane et al. described treatment periods of a median of 7 years4. The absolute risk of AFF associated with treatment with BPs is between 2 cases per 100,000 patients/year after 2 years of treatment and 78 cases per 100,000 patients/year after 8 years of treatment25. These data suggest that the duration of BP therapy would positively influence the risk of suffering these fractures. In our study, the cases of 6 patients with AFF after a long-term treatment with BPs are consistent with this association. In addition, the occurrence of AFFs in the 3 sisters suggests a genetic predisposition with a determining role in the pathology. This study has been the first exome analysis of AFF patients. We have prioritized rare, non-synonymous mutations, shared by the 3 sisters. No mutation was found in homozygosis or compound heterozygosis in any gene. These findings go against a pattern of recessive inheritance for these cases and are consistent with the fact that AFF is not a severe genetic disease that occurs during the early stages of life.
However, in the dominant model, 34 mutated genes were found, some very important for bone metabolism. In an earlier work that aimed to discover the genetic causes of AFFs, an exome chip with >300,000 known coding variants was used and 21 overrepresented rare variants were found in 13 AFF patients26. However, none of these risk alleles was found in the patients analyzed in our study. Specifically, no variants were found in the PPEF2 gene, the only one with a change significantly associated with the phenotype in the study by Pérez-Núñez et al.26 This points to a heterogeneous genetic base for AFFs. In any case, it is important to point out that our methodological approach differs from that of the aforementioned study in that we analyzed the entire exome sequence, which allowed us to find variants not previously described.
In the present study, the only gene with mutations in the 3 sisters and in unrelated patients was CYP1A1. Recently, Peris et al.27 sequenced this gene in 17 AFF patients and found another mutation in one of them. The CYP1A1 gene encodes the cytochrome P450 1A1 enzyme that is involved in the metabolism of drugs and xenobiotics. It is a hydroxylase of aryl hydrocarbons and their potential exogenous substrates include polycyclic aromatic hydrocarbons, and is involved in the formation of different human cancers. Its endogenous substrates include eicosanoids, which can generate biologically active products that act in the vascular system, among others. This gene is also responsible for the hydroxylation of 17β-estradiol, estrone and vitamin D in extrahepatic tissues28. This is consistent with its role in bone biology, an idea supported by Napoli et al.29, who demonstrated that the C4887A polymorphism was related to a significant increase in the catabolism of estrogen and a low femoral bone mineral density (BMD). postmenopausal women. Therefore, CYP1A1 is presented as another potential susceptibility gene to AFFs, although the exact mechanism of its action on bone metabolism is still unknown and more studies are needed to elucidate it.
Among the other genes with variants in the three sisters, FN1 encodes fibronectin, an extracellular matrix protein necessary for the regulation of the deposition of type I collagen by osteoblasts, essential for the mineralization of the extracellular matrix, and whose levels have been affected by treatment with BPs30. We found that the three sisters were carriers of a mutant double allele (p.V2241I and p.R1496W) in FN1, where the two mutations were considered as harmful by the predictors of pathogenicity. This altered fibronectin could affect bone mineralization and/or response to BPs and be related to the risk of AFF in these women. We also found mutated 2 regulators of small GTPases: SYDE2 and NGEF. Their respective functions (activation of RHO GTPases and exchange of their guanine nucleotides) are clues about possible effects on osteoclastic function and response to BPs. The RHO GTPases are in the path of the mevalonate in a position below the site of action of the BPs, since they have to be prenylated (farnesylated or geranylgeranylated) for their correct cellular function. On the other hand, our gene/protein interaction network shows how NGEF is closely related to ephrins and ephrin receptors (Figure 2b), which have a key role in the mechanism of coupling between osteoclasts and osteoblasts31. Another group of genes mutated in the 3 sisters encode nuclear proteins with pleiotropic effects on gene expression and/or DNA repair (KDM4C, XAB2, NVL, NKAP, ERCC6L2). Of these, we highlight the KDM4C gene, which encodes a lysine-specific demethylase that contains a JmjC domain, which has been previously associated with the age of menarche32, a biomarker for bone density.
Other genes found mutated in the sisters were the PGRMC1 gene that encodes component 1 of the progesterone membrane receptor, and which was previously associated with premature ovarian failure33; the COG4 gene (which codes for subunit 4 of the conserved oligomeric Golgi complex), which is relevant given the importance of transporting vesicles through the Golgi in osteoclasts34; and the EML1 gene (which encodes a microtubule-associated protein) that may be important in relation to the primary cilium in osteocytes35. Overall, the functions and prior knowledge of 13 of the 34 genes mutated in the 3 sisters are consistent with their possible involvement in the pathology. These mutations were searched in the 3 unrelated AFF patients, with negative results.
However, through an approach of candidate genes, mutations in these patients were found in two key proteins for bone remodeling (RUNX2 and MMP9) and in another enzyme of the mevalonate pathway (MVD, mevalonate diphosphate carboxylase). RUNX2 is an essential transcription factor for osteoblastic differentiation36, whereas MMP9 is a metalloprotease expressed in osteoclasts that degrades the extracellular bone matrix37, affecting the architecture of trabecular bone and the structure of cortical bone38. For these reasons, both may be involved in the risk to the AFF. It is known that RUNX2 activates gene expression of MMP939 and this interaction may have synergistic effects on the biomechanical properties of bone in patient AFU3, which has both mutations (Note: this interaction is not shown in Figure 2a so that other interactions can be shown clearly). Finally, in the AFU2 patient, a change mutation was found in the MVD gene, adding a second mutated protein from the mevalonate pathway. Figure 3 shows, in the context of bone cells, the proteins encoded by the genes we have found mutated and whose function in bone is known or predicted.

Figure 3 Proteins encoded by the mutated genes in AFF patients in this study and related to bone function
Taken together, all these rare variants can be part of a genetic background associated with developing bone changes that give rise to AFFs and the possible negative interaction with BPs. It is likely that several genes with small additive effects, and their interactions, are involved in AFFs related to BPs. In addition, each individual patient could be a carrier of different specific genetic variants.
The strengths of this study are the possibility of analyzing 3 sisters with AFF and the sequencing approach of the complete exome, which lacks a previous hypothesis. In this sense, we were able to identify harmful mutations in genes belonging to the mevalonate pathway, as well as other genes related to bone metabolism. On the other hand, the low number of patients and controls studied is a limitation of the study. Further studies of exome sequencing of additional AFF patients and of nonfractured patients with a long-term treatment with BPs (acting as controls) will be necessary to clarify the precise role of these genes and mutations. Despite the biological plausibility of the damaging effect of the mutations found, the replication of these findings is needed.
The identification of the genetic background for atypical fractures of the femur opens the door to the future development of tools for diagnosis and prediction of the risk of suffering this type of fracture to determine the suitability of BP treatment.
Bibliografía
01 Freemantle N, Cooper C, Diez-Perez A, Gitlin M, Radcliffe H, Shepherd S, et al. Results of indirect and mixed treatment comparison of fracture efficacy for osteoporosis treatments: A meta-analysis. Osteoporos Int. 2013;24(1):209-17. [ Links ]
02 Peng J, Liu Y, Chen L, Peng K, Xu Z, Zhang D, et al. Bisphosphonates can prevent recurrent hip fracture and reduce the mortality in osteoporotic patient with hip fracture: A meta-analysis. Pakistan J Med Sci. 2016;32(2):499-504. [ Links ]
03 Kennel KA, Drake MT. Adverse effects of bisphosphonates: Implications for osteoporosis management. Mayo Clinic Proceedings. 2009;84(7):632-7. [ Links ]
04 Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29(1):1-23. [ Links ]
05 Taormina DP, Marcano AI, Karia R, Egol KA, Tejwani NC. Symptomatic atypical femoral fractures are related to underlying hip geometry. Bone. 2014;63:1-6. [ Links ]
06 Sutton RAL, Mumm S, Coburn SP, Ericson KL, Whyte MP. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res. 2012;27(5):987-94. [ Links ]
07 Meier RPH, Lorenzini KI, Uebelhart B, Stern R, Peter RE, Rizzoli R. Atypical femoral fracture following bisphosphonate treatment in a woman with osteogenesis imperfectaa case report. Acta Orthop. 2012;83(5):548-50. [ Links ]
08 Alonso N, Soares DC, V McCloskey E, Summers GD, Ralston SH, Gregson CL. Atypical femoral fracture in osteoporosis pseudoglioma syndrome associated with two novel compound heterozygous mutations in LRP5. J Bone Miner Res. 2015;30(4):615-20. [ Links ]
09 Roca-Ayats N, Balcells S, Garcia-Giralt N, Falcó-Mascaró M, Martínez-Gil N, Abril JF, et al. GGPS1 mutation and atypical femoral fractures with bisphosphonates. N Engl J Med. 2017;376(18):1794-5. [ Links ]
10 Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754-60. [ Links ]
11 Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078-9. [ Links ]
12 McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-303. [ Links ]
13 Consortium T 1000 GP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56. [ Links ]
14 Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, et al. VARIANT: Command Line, Web service and Web interface for fast and accurate functional characterization of variants found by Next-Generation Sequencing. Nucleic Acids Res. 2012;40:W54-8. [ Links ]
15 Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073-81. [ Links ]
16 Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248-9. [ Links ]
17 Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034-50. [ Links ]
18 Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57. [ Links ]
19 Boloc D, Castillo-Lara S, Marfany G, González-Duarte R, Abril JF. Distilling a Visual Network of Retinitis Pigmentosa Gene-Protein Interactions to Uncover New Disease Candidates. PLoS One. 2015;10(8):e0135307. [ Links ]
20 Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45 (D1):D369-79; [ Links ]
21 Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808-15. [ Links ]
22 Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639-45. [ Links ]
23 Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res. 2013;41(D1):D991-5. [ Links ]
24 Yuen T, Stachnik A, Iqbal J, Sgobba M, Gupta Y, Lu P, et al. Bisphosphonates inactivate human EGFRs to exert antitumor actions. Proc Natl Acad Sci U S A. 2014;111(50):17989-94. [ Links ]
25 Brown JP, Morin S, Leslie W, Papaioannou A, Cheung AM, Davison KS, et al. Bisphosphonates for treatment of osteoporosis: expected benefits, potential harms, and drug holidays. Can Fam Physician. 2014;60(4):324-33. [ Links ]
26 Pérez-Núñez I, Pérez-Castrillón JL, Zarrabeitia MT, García-Ibarbia C, Martínez-Calvo L, Olmos JM, et al. Exon array analysis reveals genetic heterogeneity in atypical femoral fractures. A pilot study. Mol Cell Biochem. 2015;409:45-50. [ Links ]
27 Peris P, González-Roca E, Rodríguez-García SC, del Mar López-Cobo M, Monegal A, Guañabens N. Incidence of mutations in the ALPL, GGPS1 and CYP1A1 genes in patients with atypical femoral fractures. JBMR Plus. 2018; [Epub ahead of print]. [ Links ]
28 Zhou SF, Liu J-P, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev. 2009;41(2):89-295. [ Links ]
29 Napoli N, Villareal DT, Mumm S, Halstead L, Sheikh S, Cagaanan M, et al. Effect of CYP1A1 gene polymorphisms on estrogen metabolism and bone density. J Bone Miner Res. 2005;20(2):232-9. [ Links ]
30 Insalaco L, Gaudio F Di, Terrasi M, Amodeo V, Caruso S, Corsini LR, et al. Analysis of molecular mechanisms and anti-tumoural effects of zoledronic acid in breast cancer cells. J Cell Mol Med. 2012;16(9):2186-95. [ Links ]
31 Sims NA, Martin TJ. Coupling the activities of bone for mation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481. [ Links ]
32 Perry JRB, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 2014;514(7520):92-7. [ Links ]
33 Mansouri MR, Schuster J, Badhai J, Stattin EL, Lösel R, Wehling M, et al. Alterations in the expression, structure and function of progesterone receptor membrane component-1 (PGRMC1) in premature ovarian failure. Hum Mol Genet. 2008;17(23):3776-83. [ Links ]
34 Xia WF, Tang FL, Xiong L, Xiong S, Jung JU, Lee DH, et al. Vps35 loss promotes hyperresorptive osteoclastogenesis and osteoporosis via sustained RANKL signaling. J Cell Biol. 2013;200(6):821-37. [ Links ]
35 Nguyen AM, Jacobs CR. Emerging role of primary cilia as mechanosensors in osteocytes. Bone. 2013;54(2):196-204. [ Links ]
36 Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747-54. [ Links ]
37 Okada Y, Naka K, Kawamura K, Matsumoto T, Nakanishi I, Fujimoto N, et al. Localization of matrix metalloproteinase 9 (92-kilodalton gelatinase/type IV collagenase = gelatinase B) in osteoclasts: Implications for bone resorption. Lab Investig. 1995;72(3):311-22. [ Links ]
38 Nyman JS, Lynch CC, Perrien DS, Thiolloy S, O’Quinn EC, Patil CA, et al. Differential effects between the loss of MMP-2 and MMP-9 on structural and tissuelevel properties of bone. J Bone Miner Res. 2011;26(6):1252-60. [ Links ]
39 Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, Stein GS, et al. The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol. 2005;25(19):8581-91. [ Links ]
Received: June 12, 2018; Accepted: September 24, 2018
 Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
