










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
On-line version ISSN 2173-2345Print version ISSN 1889-836X
Rev Osteoporos Metab Miner vol.12 n.4 Madrid Oct./Dec. 2020 Epub Apr 05, 2021
https://dx.doi.org/10.4321/s1889-836x2020000400006
CLINICAL NOTE
Can a genetic condition be diagnosed based on phenotypic characteristics? A case of pseudohypoparathyroidism in Ecuador
1AECE Research Group, The Association of Clinical Endocrinologists of Ecuador (Ecuador)
2Endocrinology Service. Abel Gilbert Pontón Hospital. Guayaquil (Ecuador)
3Endocrinology Service. Guayaquil National Police Teaching Hospital Nº 2. Guayaquil (Ecuador)
Pseudohypoparathyroidism is a rare disease of the endocrine gland. Its diagnosis should not be dismissed when hypocalcemia is accompanied by hyperphosphatemia and high levels of parathyroid hormone even if kidney failure and vitamin D deficiency do not occur. Although genetic studies provide a definitive diagnosis, biochemical tests that show hormonal resistance and phenotypic characteristics allow us to establish a diagnosis. Literature is limited in Latin America and few cases have been described. Here we report an 18-year-old male suffering pseudohypoparathyroidism and we discuss clinical characteristics, biochemical and radiographic findings, as well as treatment.
Key words Albright's hereditary osteodystrophy; parathyroid hormone resistance; pseudohypoparathyroidism; inactivating PTH/PTHrP signaling disorder; hypocalcemia; brachydactyly; Ecuador
INTRODUCTION
Pseudohypoparathyroidism (PHP) is a heterogeneous group of disorders which share in common a parathyroid hormone resistance (PTH).
Globally, estimated prevalence is 0.79/100,0001, though it depends on the analysed type of PHP, and it oscillates between 6.7 and 3.3 cases per million inhabitants in Italy2 and Japan3 respectively. Between 2000 and 2019, 325 cases4 have been described in worldwide literature, most of them in developed countries, in which in addition, PHP subtypes have been documented throughout genetic studies. 1a subtype is the most common, representing 70% of the cases1. 47 cases have been reported in Latin America between 2000 and 20205-10, the most frequent subtype being 1b, followed by 1a and 1c. Due to a lack of genetic studies, in some cases is not possible to precisely ascertain the belonging to one or another subtype10.
The following is a description of an 18-year-old male’s clinical case presenting phenotypical features of Albright's hereditary osteodystrophy (AHO).
CASE REPORT
An 18-year-old man attended an endocrinology consultation after being referred by the neurology service due to seizures associated with persistent hypocalcaemia.
He presented a history of generalized tonic-clonic seizures from 15 years of age, for which he had been previously hospitalized, showing hypocalcaemia, treated with intravenous calcium and anticonvulsants to control the emergency and oral calcium supplements upon discharge from hospital. No further studies were carried out to determine the cause of hypocalcaemia.
His parents and first-degree relatives have no significant medical history. The patient is the child of a non-consanguineous marriage. He was born pre-term due to premature rupture of membranes at 35 weeks of gestation, with neonatal hypoxia and hypotonia. He presented late motor and language development, requiring language therapy from 5 to 7 years of age and physiotherapy since he was 2, in addition to school learning tardiness and permanent overweight. At 12 years of age he was diagnosed with primary hypothyroidism and since then he has taken 150 µg of levothyroxine sodium.
The patient is the youngest of 3 siblings who have no significant medical history.
On physical examination, he presented a characteristic phenotype: obese, short height, round face, prominent forehead, low nasal bridge, short neck, brachydactyly, and incomplete teeth (Figure 1). His weight was 68.8 kg, and his height, 153 cm (<3rd percentile); the body mass index was 34 (>97th percentile). His hands and feet were small, the first finger of the two hands being substantially small by far, characteristic corresponding to type E2 brachydactyly. The X-ray shows marked metacarpal shortening and that of the distal phalanx of the thumbs (Figure 2). In addition, subcutaneous calcifications are detected radiologically in the posterior thorax and dorsum of feet.
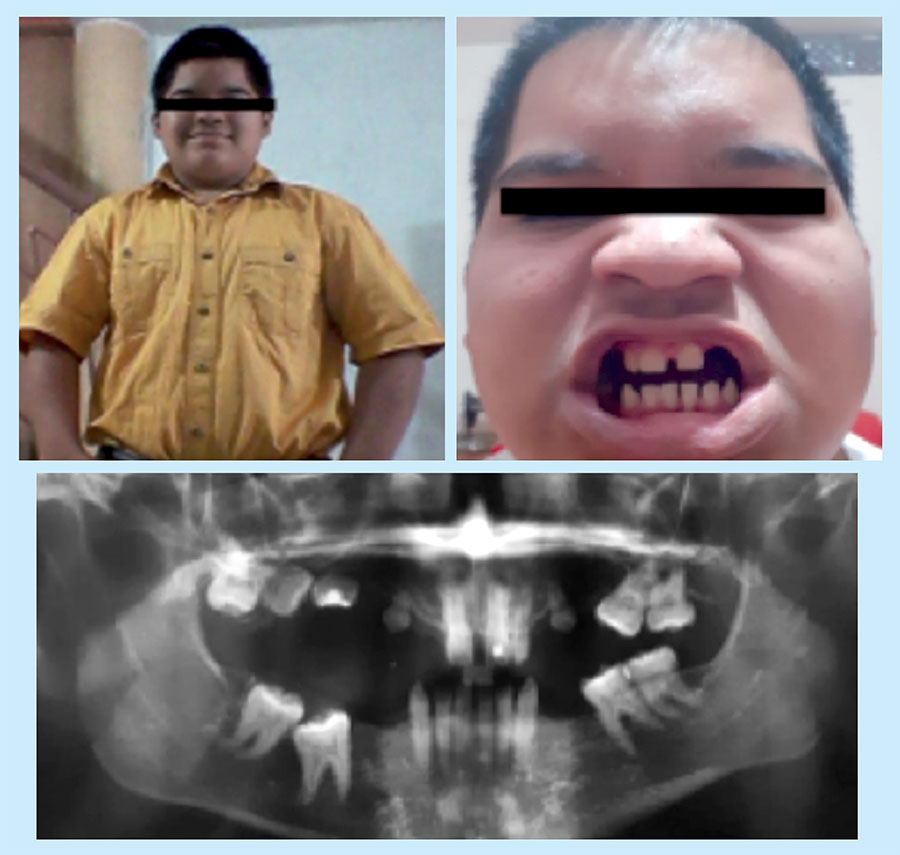
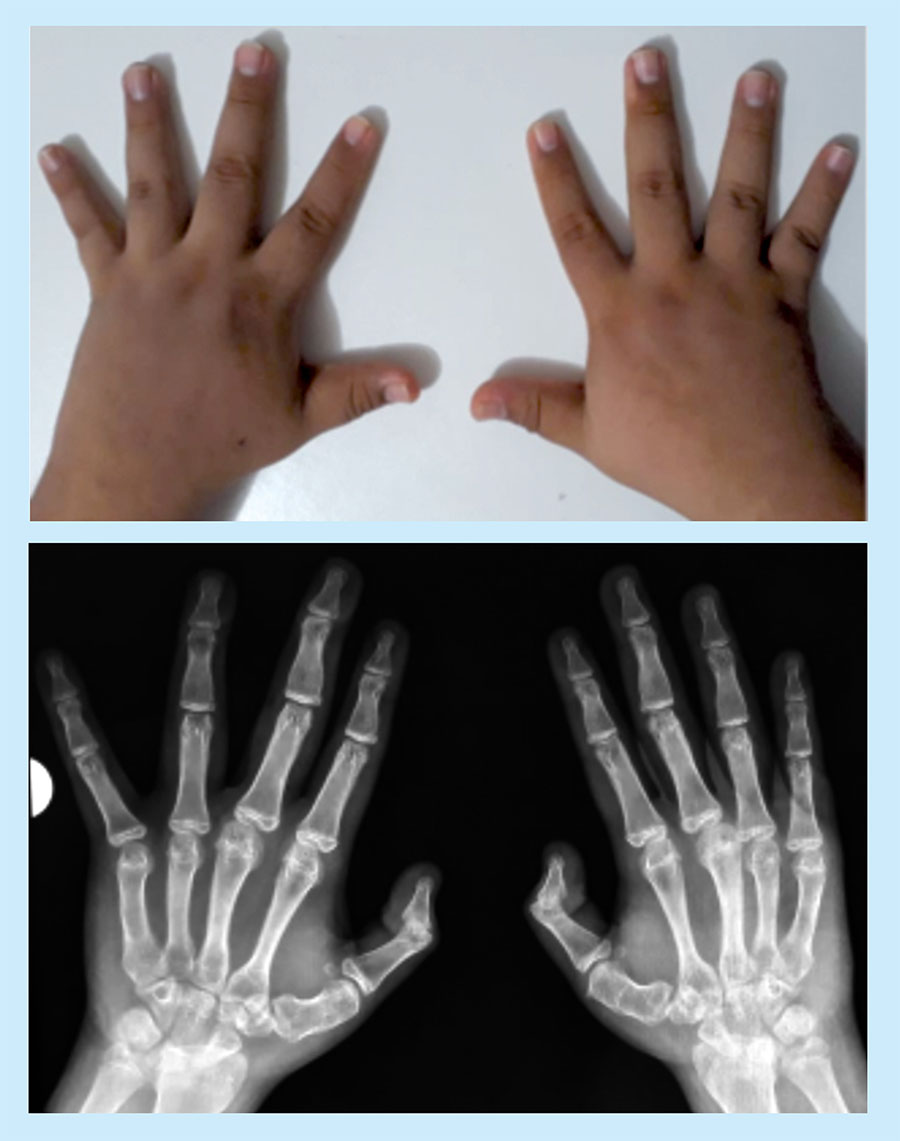
Figure 2 Photograph and radiography of the hands. Marked shortening of the metacarpal bones and distal phalanx of the thumbs
The pubertal stage was Tanner V: penis length, 15 cm (normal >15 cm); testicular volume, 30 ml bilaterally (normal >20 ml).
Chvostek and Trousseau signs were positive, with total calcium levels: 1.67 mmol / L (normal value: 2.12-2.57 mmol/L); serum phosphorus: 1.81 mmol/L (normal value: 0.8-1.6 mmol/L); parathyroid hormone (PTH): 12.05 pmol/L (normal value: 1.5-8.97 pmol/L), and total vitamin D (25-OH-D): 76.38 nmol/L (normal value: 25- 137 nmol/L).
The biochemical evaluation established that the seizures, paresthesia, and Chvostek and Trousseau signs were attributed to hypocalcaemia. He was treated with an intravenous calcium infusion and later referred to the endocrinology department for a comprehensive evaluation.
In dual energy X-ray absorptiometry (DXA) bone densitometry, with DEXXUM-T equipment (OsteoSys - Seoul, Korea), he presented a decrease in bone mass for his age and gender in the lumbar area (L1-L4) , with preservation of bone mass in the femoral neck (lumbar spine: 1.044 g/cm2, Z-score: -2.6; and femoral neck: 1.146 g/cm2, Z-score: 0.3).
The computed tomography of the skull revealed cortical and subcortical calcifications, peri-ventricular, nuclei of the base and in the cerebellum. The nuclear magnetic resonance study with contrast medium showed periventricular calcifications.
The results of the current hormonal and biochemical determinations are presented in table 1. Total vitamin D (25 hydroxyvitamin D) was determined by electrochemiluminescence (normal value: 25-137 nmol/L). PTH was determined by chemiluminescence (normal value: 1.5-8.97 pmol/L). The determination of 1.25 dihydroxyvitamin D and PTH-induced cAMP was not carried out due to unavailability of hospital tests and financial constraints.
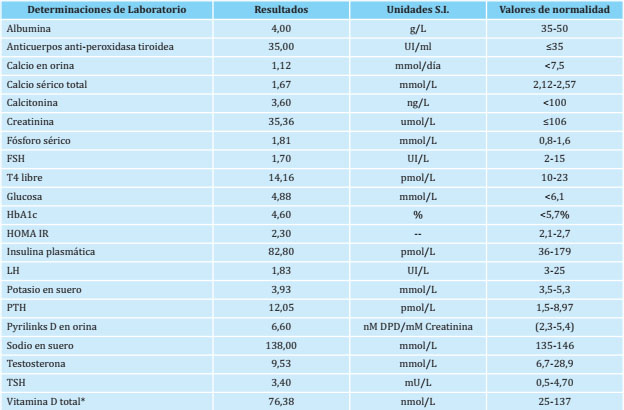
The complete blood count, blood glucose levels and liver and kidney function tests were normal. The biochemical and hormonal study of his mother shows normal values: calcium 2.35 mmol/L, phosphorus: 1.13 mmol/L, TSH: 3.5 mU/L, free T4: 14.1 pmol/L, PTH: 2.58 pmol/L.
At follow-up, serum calcium levels and urinary calcium/creatinine ratio were monitored to achieve optimal serum calcium levels.
The patient is currently being treated with oral calcium carbonate supplements, 3 g/day; oral calcitriol, 1.5 mg/day; vitamin D3, 2,000 IU/day, and levothyroxine, 150 µg/day. He attends outpatient check-ups every 3 months were he is taken measurements of serum calcium and phosphorus, PTH, vitamin D and thyroid hormones. In addition, he regularly attends the Neurology Service to control his seizures and the Psychology Department to get support for him and his family. The Nutrition Service offers dietary advice, and due to her dental alterations he is under permanent dental treatment.
DISCUSSION
PHP is a clinically dysmorphic syndrome characterized by skeletal and developmental defects11, including short height, rounded face, short fourth metacarpal bones as well as other bones in the hands and feet, obesity, dental hypoplasia, and soft tissue calcifications or ossifications12,13. However, some cases may present unusual phenotypic characteristics12,13. The biochemical characteristics of patients with PHP are hypocalcaemia, hyperphosphatemia and elevated levels of PTH12.
In the present case, the diagnosis of pseudohypoparathyroidism was considered given the laboratory results compatible with resistance to PTH (hypocalcaemia, hyperphosphatemia and elevated PTH), together with the phenotypic characteristics of Albright's hereditary osteodystrophy14 (AHO), which guided us towards a 1a or 1c PHP type.
Hypocalcaemia is a consequence of PTH bone resorption response loss, resulting in a defective mobilization of calcium from the bone and a lower absorption of calcium in the intestine12.
Brachydactyly, described as shortening of the III-V metacarpal/metatarsal bones and the distal phalanx of the first finger, is one of the most specific characteristics of the Albright phenotype14. From the phenotypic characteristics of AHO, we highlight PHP type E brachydactyly; and this patient presents significant shortening in the metacarpus and distal phalanx of the thumb in both hands that could be considered a type E2 brachydactyly14,15.
There is an association of PHP with variable resistance to multiple hormones that act through the Gsα protein. Resistance to TSH is the hormonal alteration that has been most commonly associated and can even be diagnosed before the appearance of phosphocalcium metabolism disorders16. In this case, the absence of antithyroid antibodies supports the resistance to TSH diagnosis11.
Reproductive dysfunction has been associated with 1a PHP; however, the effects of hypogonadism are less evident in men16. The normal secondary sexual characteristics and the determination of sex hormones rule out the possibility of alteration in the gonadal axis in our patient.
There are discrepancies on the effects of PHP on the skeleton8,17,18. Some studies have reported that bone density is reduced in patients with PHP17. However, Long et al. analysed the bone mineral density in 22 subjects with 1a PHP and found that bone mass was normal or increased in all the studied bone regions18. On the contrary, in this case the bone mineral density measured in the lumbar region shows decreased values compared to the controls of same age and gender, with preservation of bone mass in the femoral neck.
Between 2000 and 2019, the international literature described approximately 325 cases of PHP4,16,17. A series of 60 PHP cases was published in Denmark in 2016, but only 30 (50%) of them underwent genetic testing for PHP, of which in 14 a mutation in the GNAS1 gene was identified. In those which could not be genetically confirmed (76%), characteristic biochemical and hormonal criteria were accepted as diagnosis, excluding cases with confirmed evidence of kidney failure, vitamin D deficiency, or any other known cause of secondary hyperparathyroidism17. In 2013, in a series of 72 cases with PHP treated in the Spanish National Health System, genetic confirmation could be made in 63 of the cases (88%)16.
In Latin America, after a search throughout the literature ranging from the years 1957 and 2020, we found 32 publications, in which 56 cases of PHP are reported. Genetic studies in order to confirm the diagnosis were performed in only 6 of these publications. In most cases the diagnosis was based on the biochemical/hormonal and phenotypic profile.
There are no previously reported cases in our country, which is possibly due to subdiagnosis. However, data published worldwide describe the phenotypic characteristics associated with the biochemical alteration compatible with the findings in our patient. In our case, the clinical diagnosis of PHP could not be confirmed by a genetic study, but, as it is accepted in the literature, clinical and biochemical evidence are sufficient to precise the diagnosis of PHP4,11.
In accordance with international criteria12,15, our aim in long-term treatment has been to reduce the serum PTH level to the upper level of the reference range with 1-25 dihydroxyvitamin D and oral calcium, to improve calcium reabsorption in the distal renal tubule, prevent hypercalciuria and avoid alterations in bone mineralization18.
CONCLUSIONS
In Latin American countries in which genetic studies are not available, we must bear in mind that, when presented with a patient with severe and persistent hypocalcaemia associated with high PTH, normal kidney function and a characteristic phenotype, the suspicion of a PHP must arise, even despite the lack of genetic confirmation.
Bibliografía
1 Mantovani G, Linglart A, Garin I, Silve C, Elli FM, de Nanclares GP. Clinical utility gene card for: pseudohypoparathyroidism. Eur J Hum Genet. 2013;21(6). [ Links ]
2 Mantovani G. Pseudohypoparathyroidism. Orpha.net. Available at http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=12935&Disease_Disease_Search_diseaseGroup=Pseudohypoparathyroidism&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Pseudohypoparathyroidism&title=Pseudohypoparat. October 2014; Accessed: August 29, 2020. [ Links ]
3 Nakamura Y, Matsumoto T, Tamakoshi A, Kawamura T, Seino Y, Kasuga M, et al. Prevalence of idiopathic hypoparathyroidism and pseudohypoparathyroidism in Japan. J Epidemiol. 2000;10 (1):29-33. [ Links ]
4 Mantovani G, de Sanctis L, Barbieri AM, Elli FM, Bollati V, Vaira V, et al. Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of Albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010;95(2):651-8. [ Links ]
5 Peña C, Pinochet C, Florenzano P, Mendoza C, Garfias C, Aracena M, et al. Pseudohypoparathyroidism report of two cases of late presentation. Rev Med Chile. 2018;146:116-21. [ Links ]
6 Reis MT, Matias DT, Faria ME, Martin RM. Failure of tooth eruption and brachydactyly in pseudohypoparathyroidism are not related to plasma parathyroid hormone-related protein levels. Bone. 2016;85:138-41. [ Links ]
7 Trejo MC, Román-González A, Ruiz S, Tobon C, Castano P, Arango C, et al. Late diagnosis of pseudohypoparathyroidism in adulthood. Case series. Rev Fac Med. 2018;66(4):643-9. [ Links ]
8 Fernandez M, Zambrano MJ, Riquelme J, Castiglioni C, Kottler ML, Jüppner H, et al. Pseudohypoparathyroidism type 1B associated with assisted reproductive technology. J Pediatr Endocrinol Metab. 2017;30(10):1125-32. [ Links ]
9 Arús Fernández AE, González Fabian L, García Bacallao E, Cormac Tur JB, González Fernández P, Castaño González L, et al. Seudohipoparatiroidismo 1b. Rev Cubana Endocrinol. 2019:30(2); e173. [ Links ]
10 Carvalho B, Nascimento I, Cunha C, Morais J, Carvalho L. Osteodistrofia Hereditária de Albright: Um Relato de Caso. Rev Ciênc Saúde Nova Esperança. 2020;18(1):41-8. [ Links ]
11 Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Usardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. 2018; 14(8):476-500. [ Links ]
12 Linglart A, Levine M.A, Jüppner H. Pseudohypoparathyroidism (Chapter 86.). In: John P. Bilezikian, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Ninth Edition. Hoboken, New Jersey: Wiley-Blackwell; 2019. 661-673. [ Links ]
13 Hejlesen J, Underbjerg L, Gjørup H, Sikjaer T, Rejnmark L, Haubek D. Dental anomalies and orthodontic characteristics in patients with pseudohypoparathyroidism. BMC Oral Health. 2019; 20(1):2. [ Links ]
14 David A, Vincent M, Quéré MP, Lefrançois T, Frampas E, David A. Isolated and syndromic brachydactylies: Diagnostic value of hand X-rays. Diagn Interv Imaging. 2015;96(5):443-8. [ Links ]
15 Germain-Lee EL. Management of pseudohypoparathyroidism. Curr Opin Pediatr. 2019;31(4):537-49. [ Links ]
16 Fernández-Rebollo E, Lecumberri B, Gaztambide S, Martinez-Indart L, Perez de Nanclares G, Castaño L; Spanish PHP Group. Endocrine profile and phenotype-(epi)genotype correlation in Spanish patients with pseudohypoparathyroidism. J Clin Endocrinol Metab. 2013;98(5):E996-1006. [ Links ]
17 Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Pseudohypoparathyroidism - epidemiology, mortality and risk of complications. Clin Endocrinol (Oxf). 2016;84:904–11. [ Links ]
18 Long DN, Levine MA, Germain-Lee EL. Bone mineral density in pseudohypoparathyroidism type 1a. J Clin Endocrinol Metab. 2010;95(9):4465-75. [ Links ]
Received: September 25, 2020; Accepted: December 03, 2020
 Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
