










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.13 no.1 Madrid ene./mar. 2021 Epub 17-Mayo-2021
https://dx.doi.org/10.4321/s1889-836x2021000100004
ORIGINALS
Search for variants of the LRP4 gene in women with high bone mass and in patients with Chiari type I malformation
1aGenetics, Microbiology and Statistics Department. Biology Faculty. University of Barcelona. Barcelona (Spain)
1bCenter for Biomedical Research of the Rare Diseases Network (CIBERER). (Spain)
1cInstitute of Biomedicine of the University of Barcelona (IBUB). Barcelona (Spain)
1dSan Joan de Déu Research Institute (IRSJD). Barcelona (Spain)
Objetive
LRP4 is an essential facilitator in sclerostin-specific inhibition of the canonical Wnt pathway. Mutations in LRP4 have been associated with various conditions, including bone growth disease, sclerosteosis, and Chiari type I malformation (CMI).
Material and methods
The LRP4 has been re-sequenced in two patient cohorts with high bone mass phenotype (HBM) and with CMI aimed at finding causal variants.
Results
Among the mutations found, we would highlight: 1) a missense mutation in a patient with CMI, which does not co-segregate with the phenotype in the family; 2) a previously undescribed intronic mutation (c.3364+16A>C) in a woman with HBM; and 3) an intronic mutation in a woman with HBM with a very low frequency in the European control population.
Conclusion
Although we have not found variants in LRP4 to explain the HBM or CMI phenotype in the patients studied, we encourage other researchers to analyze the LRP4 gene in their patients as it is a good functional candidate for both phenotypes.
Key words LRP4; HBM; Chiari malformation type I; bone mineral density; sclerostin
INTRODUCTION
The Wnt signalling pathway is involved in a wide range of processes, including bone development and homeostasis1. In accordance with this, mutations have been identified in various components of the Wnt pathway that cause different musculoskeletal diseases2. The canonical Wnt pathway begins with the formation of a heterotrimeric complex between a co-receptor, LRP5/6, a ligand, WNT, and a receptor, FZD, which produces an accumulation of β-catenin that, once in the nucleus will activate the transcription of numerous important target genes for bone1. This activation is finely regulated by a series of extracellular inhibitors such as DKK1 and sclerostin that bind to LRP5/6, preventing the formation of the heterotrimeric complex. For DKK1 and sclerostin to exert their inhibitory activity, they must form another heterotrimeric complex with LRP5 and KREMEN1/2 or LRP4, respectively. Although in the case of DKK1 the presence of KREMEN does not seem to be necessary to carry out a correct inhibition, the presence of LRP4 is essential for the inhibitory function of sclerostin3,4.
LRP4 mutations have been described in humans which cause different diseases that affect not only bone mass, but also the regulation of the extremities and kidneys among other, depending on the position of where the mutation occurs2. Specifically, mutations in the central cavity of the third β-propeller cause sclerosteosis, characterized by variable syndactyly and progressive bone overgrowth, particularly severe in the facial skeleton and skull. These patients present an increase in the Wnt pathway in osteoblasts, generating an increase in bone formation4-6. In addition to these mutations, in 2017, Merello et al.7 described a mutation in the second β-propeller domain (p.Thr851Arg) that cosegregated with the phenotype in a family with type I Chiari malformation (CMI). This is a malformation of the central nervous system characterized by a caudal displacement of the cerebellar tonsils, which generates varied symptoms both in onset and in severity (Orphanet, https://www.orpha.net/consor/cgi-bin/ index.php). Although some patients with CMI may be asymptomatic, others can present with a suboccipital headache and neck pain, among others. It is interesting to note that this malformation was described in a patient who presented a phenotype of high bone mass (HBM) due to gain-offunction mutations in LRP5, thus suggesting a possible relationship between CMI, the HBM phenotype and Wnt pathway8. Taking into account the role of LRP4 on the Wnt pathway and the important role that this pathway has in determining bone mineral density (BMD) and in the development of the skull, we hypothesized that mutations in LRP4 could be the cause of the HBM in women with this phenotype or of causing disease in patients with CMI. In this study, we have carried out a re-sequencing of the LRP4 gene in a cohort of 10 women with the HBM phenotype and in a cohort of 12 patients with CMI.
MATERIAL AND METHODS
Cohorts studied
Sarrión et al. describe the HBM cohort, in a study of 10 women with HBM phenotype9, in which this phenotype is defined as the sum of the Z-score values of the lumbar spine and femoral neck are equal or greater than 4.
The CMI cohort studied here consists of 12 patients with unrelated CMI, diagnosed and treated at the Hospital del Mar in Barcelona. The diagnosis of CMI is based on the position of the cerebellar tonsils by brain magnetic resonance imaging (Achieva 3.0 T, Philips, Amsterdam, The Netherlands) with a herniation equal to or greater than 5 mm on a mid-sagittal T1-weighted image in the presence of signs or symptoms indicating neural compression at the cranio-vertebral junction, syringohydromyelia, cerebellar dysfunction or intracranial hypertension. In addition, information is available on 8 relatives of 4 of the patients with CMI.
LRP4 re-sequencing
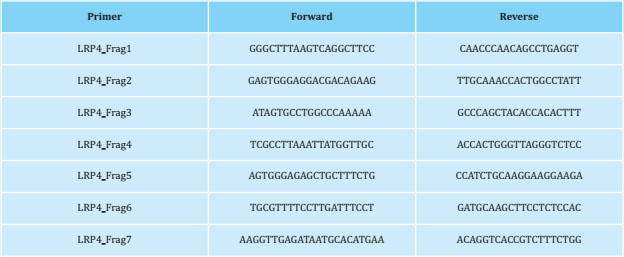
Genomic DNA from CMI cases and their relatives and from HBM women was isolated. women was isolated from peripheral blood leukocytes using the Wizard® Genomic DNA Purification Kit (Promega, Madison, Wisconsin, USA), according to with the manufacturer's instructions. Regarding the re-sequencing, specifically, we have amplified the exons that code for the mutations that cause Cenani-Lenz syndrome (p.D137N, p.C160Y, p. D449N, p. T461P, p.L473F, p. D529N, p.L953P, p.C1017R, p.R1277H, p.E1233K) and the third β-propeller domain where the mutations causing myasthenic syndrome and sclerosteosis are located. Amplification of each of these fragments was done by polymerase chain reaction (PCR) using GoTaq Flexi DNA polymerase (Promega). The PCR fragments were analyzed by agarose gel electrophoresis method and their purification was done on MultiScreen™ Vacuum Manifold 96-well plates (Merck Millipore). The purified PCR products were sequenced using the Sanger method in the genomics service of the CCiTUB (Genómica, Parc Científic, Barcelona, Spain). The labeling kit used was BigDye™ Terminator v3.1 Cycle Sequencing Kit (ThermoFisher), detection and electrophoresis were carried out on the 3730 Genetic Analyzer and 3730xl Genetic Analyzer (Thermo-Fisher) automatic capillary sequencers models. The design of the primers was based on the consensus sequence ENSG00000134569 (LRP4; GRCh37.p13). Their sequence is presented in table 1.
Bioinformatic analysis and in silico predictions of the effect of the variants
To identify and characterize all the variants, we have used information culled from the Ensembl database GRCh37.p13 and ENCODE. The minor allele frequency (MAF) of each of the variants is extracted from the non-Finnish European population of gnomAD v2.1.1. We have used SIFT, (http://sift.bii.a-star.edu.sg/) and Poly-Phen (http://genetics.bwh.harvard.edu/pph2/) to test the effect of the change variants of amino acid (missense). The GTEx database (www.gtexportal.org/home/) has been used to identify variants that act as eQTLs.
RESULTS
Re-sequencing of LRP4 in women with HBM phenotype and in patients with CMI
In the LRP4 re-sequencing have identified 12 variants, of which one was not previously described (c.3364+16A>C; table 2). This variant has been found in heterozygosity in the HBM2 woman who presents a Zscore sum (lumbar spine and femoral neck) of 4.6. Six of the 12 variants identified are found both in the cohort of women with the HBM phenotype and in the cohort of patients with CMI, with a similar frequency in both cohorts to that of the European population. Furthermore, we have found 4 variants only present in the HBM cohort and 2 variants only present in the CMI cohort. All the variants described, except rs558515201 and the variant c.3364+16A>C, appear as eQTL of different genes and tissues in the GTEx database. Specifically, the rs17790156, rs61898529, rs2306028, rs964551 and rs2306032 variants are LRP4 eQTL.
Table 2. Variants found in LRP4 re-sequencing in women with HBM phenotype and in patients with CMI. The genotypes in the first column are indicated by the nucleotides of the coding strand of the LRP4 gene, which is the reverse of that which is used in a standard way to indicate genomic SNVs. For this reason, for example, the A allele of c.431-12G> A is equivalent to the T allele of rs139371503, and the same for the other variants

MAF: minority allele frequency; EUR: non-Finish European population of gnomAD v2.1.1.; HBM: high bone mass phenotype; CMI: Chiari type I mal-formation; dup: dup: duplication; M: missense variant; I: intronic variant; S: synonymous variant; eQTL: described as eQTL in Gtex; T: tolerated by SIFT; B: benign by Polyphen-2.
DISCUSSION
Different studies have highlighted the role of the LRP4 gene in determining BMD, as mutations in it generate a phenotype of bone overgrowth. Furthermore, it has also been associated with CMI in a report on whole exome sequencing7. In the study presented here, we have re-sequenced the regions of the gene that contain mutations associated with different conditions. In it, we have only found a missense mutation in LRP4 in a patient with CMI that does not co-aggregate with the phenotype in the family, thus ruling out its ability to cause the disease (Figure 1). It is interesting to note that we have found a variant not previously described (c.3364+16A>C) and the variant rs558515201, which has a very low frequency in the European population (MAF=0.00006480), in heterozygosity in two women with HBM. Furthermore, the rs3751097 and rs540384558 variants, present in both HBM and CMI patients, are found in a cis regulatory element categorized as a distal enhancer-like signature by ENCODE. On the other hand, contrary to expectations, we found in both cohorts a slightly lesser frequency for the minor allele of the rs2306032 variant which has been defined as a protector in whole genome association study with total BMD10.
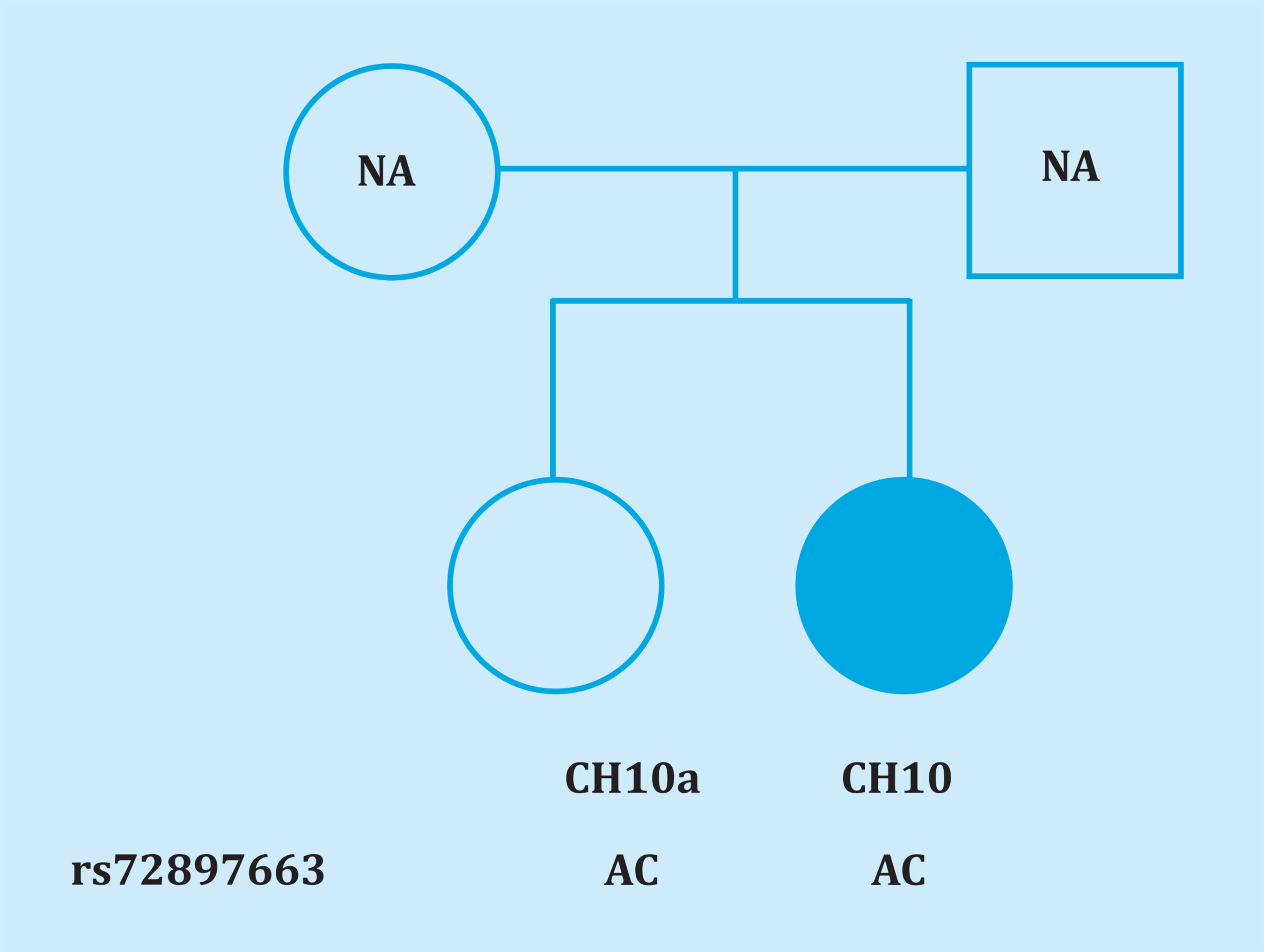
Figure 1. Pedigree of the family of the CMI CH10 patient. NA: no data on the parents. Genotypes are indicated with the nucleotides of the coding strand of the LRP4 gene, which is the reverse of that used in a standard way to indicate genomic SNVs. Therefore, allele C is equivalent to allele G of rs72897663
The low number of patients must be taken into account as a limitation of our study. This could explain both the absence of causal variants in the LRP4 gene in these two cohorts and the results contrary to those expected in allele frequency in the SNP rs2306032. Thus, increasing the size of the cohorts would provide us with a deeper understanding of the role of LRP4 on these phenotypes.
In conclusion, although LRP4 is undoubtedly an important gene for bone biology, we have not found any variant that could explain the HBM or CMI phenotype. Despite these negative results, it is interesting to consider LRP4 in the selection of candidate genes that may explain HBM or CMI-causing phenotypes.
Bibliografía
1 Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013:19(2):179-192. [ Links ]
2 Huybrechts Y, Mortier G, Boudin E, Van Hul W. WNT signaling and bone: lessons from skeletal dysplasias and disorders. Front Endocrinol (Lausanne). 2020;11:165. [ Links ]
3 Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, et al. Kremen proteins are dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417(6889):664-667. [ Links ]
4 Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286 (22): 19489-19500. [ Links ]
5 Fijalkowski I, Geets E, Steenackers E, Van Hoof V, Ramos FJ, Mortier G, et al. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J Bone Miner Res. 2016; 31(4):874-881. [ Links ]
6 Bukowska-Olech E, Sowińska-Seidler A, Szczałuba K, Jamsheer A. A novel biallelic splice-site variant in the LRP4 gene causes sclerosteosis 2. Birth Defects Res. 2020;112(9):652-659. [ Links ]
7 Merello E, Tattini L, Magi A, Accogli A, Piatelli G, Pavanello M, et al. Exome sequencing of two italian pedigrees with non-isolated Chiari malformation type i reveals candidate genes for cranio-facial development. Eur J Hum Genet. 2017;25(8):952-959. [ Links ]
8 Whyte MP, Reinus WH, and Mumm S. High-bone-mass disease and LRP5. N Engl J Med. 2004;350(20):2096-2109. [ Links ]
9 Sarrión P, Mellibovsky L, Urreizti R, Civit S, Cols N, García-Giralt N, et al. Genetic analysis of high bone mass cases from the BARCOS cohort of Spanish postmenopausal women. PloS One. 2014;9(4):e94607. [ Links ]
10 Medina-Gomez C, Kemp JP, Trajanoska K, Luan J, Chesi A, Ahluwalia TS, et al. Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am J Hum Genet. 2018;102(1):88-102. [ Links ]
Received: December 22, 2020; Accepted: March 01, 2021
 Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
