










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.14 no.2 Madrid abr./jun. 2022 Epub 12-Sep-2022
https://dx.doi.org/10.4321/s1889-836x2022000200001
LETTER TO THE EDITOR
Familial hypocalciuric hypercalcemia. Concerning two cases
1Endocrinology and Nutrition Department. General University Hospital of Ciudad Real (Spain)
2 Internal Medicine Service. HGUCR. General University Hospital of Ciudad Real (Spain)
3 Endocrinology and Nutrition Service. Salamanca University Hospital Complex (Spain)
Familial hypocalciuric hypercalcemia (FHH) is a syndrome characterized by the association of mild or asymptomatic hereditary hypercalcemia and hypocalciuria. 3 subtypes have been described (FHH1, FHH2 and FHH3). FHH1, the most common, is due to inactivating mutations in the calcium-sensitive receptor (CaSR) gene1-3. Its prevalence is low, the inheritance is autosomal dominant, and it is often diagnosed by chance, because it is rarely symptomatic. Due to its clinical benignity, it is essential to establish a differential diagnosis (DD) with primary hyperparathyroidism (PHPT) to avoid unnecessary examinations and treatments. Routine genetic testing is not accurate because biochemical tests usually establish the diagnosis4.
Two cases of FHH1 are described. In one, the need for a genetic study is debatable. In the other, the mutation found had not been previously described.
Table 1. Biochemical data of the cases
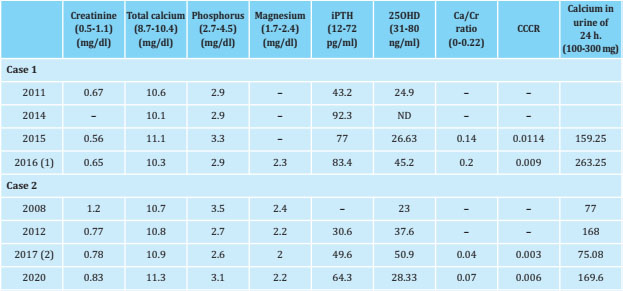
iPTH: intact parathormone; 25OHD: 25‐OH vitamin D; CCCR: calcium/creatinine clearance ratio (index). (1) and (2) at that time were on calcifediol treatment.
The first, a 47-year-old woman, consulted for mild hypercalcemia. With no relevant medical history or semiology of hypercalcemia, she had mild hypercalcemia with normal intact parathyroid hormone (iPTH), but the diagnostic study was not completed due to non-appearance during the following three years. Referred again by elevated iPTH, tests were requested to rule out PHPT. The results confirmed the persistence of hypercalcemia with slightly elevated iPTH and normal vitamin D, but without hypercalciuria (urine calciuria 24 hours 159.25 mg/24) (table 1). The cervical ultrasound and scintigraph scan did not reveal pathological data at the parathyroid level. The patient reported, at that time, that her mother and 2 of her 6 siblings had FHH due to a mutation in the CaSR gene. The genetic study of the patient confirmed the existence of the same CaSR mutation as her relatives: change c.1394G>A; P. (ArgRG465Gln).
The second is a 36-year-old man referred for hypercalcemia, treated with oral corticosteroids (dexamethasone 0.5 mg/day, in recent years) for non-classical congenital adrenal hyperplasia. No other personal or family history of interest or semiology was attributable to hypercalcemia. He had mild hypercalcemia, a normal iPTH concentration, and a “normal” 24-hour calciuria (low for the calcaemia level), which has remained practically stable, with some clear hypocalciuria, during follow-up (table 1). He was diagnosed with lumbar osteopenia, which was considered secondary to chronic corticosteroid therapy. The genetic study, carried out in 2013, found a genetic mutation c.164dupC (p. Glu56Glyfs * 9) in exon 2 of CASR in heterozygosity. This alteration had not been described up to that date.
It is very uncommon for patients with FHH to present the most common symptoms in other hypercalcemic syndromes, even when the calcemia is higher. While slight elevations in bone turnover markers can be detected, this does not affect bone mineral density or increase the incidence of fractures. Hypercalcemia in patients with FHH is barely elevated, although in some family groups it can exceed 12 mg/dL, due to the peculiarities of the mutation present in CaSR5, it is already present at birth, unlike PHPT, and persists throughout life.
Typically iPTH is inappropriately normal for calcium concentration, but occasionally it may be significantly elevated. In this case, DD with a PHPT is difficult. The other defining characteristic of the disease is excessive tubular calcium reabsorption despite hypercalcemia, which translates into a calcium/creatinine clearance ratio (CCCR) of less than 0.01 in 80% of cases. Most PHPT have a higher index (> 0.02)6. These low clearance rates in FHH persist even after complete parathyroidectomy, suggesting that calcium reabsorption is independent of PTH. CCCR has been proposed as a simple diagnostic test for a rapid DD between FHH and PHPT, taking as a cutoff point for FHH a value <0.02; but low CCCR values (between 0.01 and 0.02) have been observed in some typical PHPT, especially in those who concomitantly present with hypovitaminosis D or renal failure7.
Therefore, genetic analysis continues to be the “gold standard” test to establish this DD. The genetic study is widely accepted for those patients with a CCCR <0.028,9, although some also limit the indications to children under 10 years of age with hypercalcemia and elevated or normal PTH, atypical cases that do not present hypocalciuria or with a phenotype of FHH with normocalcemic parents (de novo CaSR mutation), cases in which there are other relatives with hypercalcemia with no known cause and when there are no family members available for testing10. The indication for the genetic study is not always easy, as shown in the first case, in which the family history would have allowed a reliable diagnosis to be established with biochemical tests, but the repeated detection of elevated iPTH and the absence of hypocalciuria influenced the decision to perform genetic analysis.
Bibliografía
1 Tellam JJ, Abdulrasoo G, Ciin LCH. Think twice: a rare calcium sensing receptor mutation and a new diagnosis of familial hypocalciuric hypercalcaemia. Endocrinology, Diabetes and Metabolism. Case repots. ID: 20-0004; June 2020 DOI: 10.1530/EDM-200004. [ Links ]
2 García-Castaño A, Madariaga L, Azriel S, et al. Identification of a novel large CASR deletion in a patient with familial hypocalciuric hypercalcemia. Endocrinology, Diabetes and Metabolism. Case repots. ID: 18-0114; December 2018 DOI: 10.1530/EDM-18-0114. [ Links ]
3 Vahe C, Benomar K, Espiard S, et al. Diseases associated with calcium-sensing receptor. Orphanet Journal of Rare Diseases (2017) 12:19. DOI: 10.1186/s13023-017-0570-z. [ Links ]
4 Olivar Roldán J, Pavón de Paz I, Iglesias Bolaños P, Montoya Álvarez T, Fernández Martínez A, Monereo Megías S. Hipercalcemia hipocaciúrica familiar: a propósito de tres casos en una misma familia. Endocrinol Nutr. 2008; 55(6):267-269. [ Links ]
5 Díaz R, Brown EM. Familial hypocalciuric hypercalcemia and other disorders due to calcium-sensing receptor mutations. En DeGroot LJ and Jameson JL, Ed. Endocrinology. Fifth edition. Philadelphia, Elsevier Saunders, 2006: pag 1595-1609. [ Links ]
6 Merino M, Vega B, Guijarro G, Navea C, Torán C, Civantos S. Hipercalcemia hipocalciúrica familiar: a veces no es lo que parece. Rev Osteoporos Metb Miner 2015;7(1):20-22. [ Links ]
7 Gorvin CM. Molecular and clinical insights from studies of calcium-sensing receptor mutations Journal of molecular endocrinology 2019;63: R1-R16. DOI: https://doi.org/10.1530/jme-10-0104.https://doi.org/10.1530/JME-19-0104. [ Links ]
8 Tosur M, López ME, Paul DL. • Primary hyperparathyroidism vs familial hypocalciuric hipercalcemia. Ann Pediatr Endocrinol Metab. 2019;24:195-198. https://doi.org/10.6065/apem.2019.24.3.195. [ Links ]
9 Christensen SE, Nissen PH, Vestergaard P, et al. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin Endocrinol (Oxf) 2008;69:713-720. [ Links ]
10 Andrade Navarro MT, Pérez González E, Cantos Pastor V, Marín Patón M, Lara Ruíz A. Hipercalcemia hipocalciurúrica familiar. Descripción de un caso. Arch Argent Pediatr 2018;116(6): e757-e761. [ Links ]
Received: July 23, 2021; Accepted: January 11, 2022
 Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
Este es un artículo publicado en acceso (Open Access) abierto bajo la licencia Creative Commons Attribution Non-Commercial, que permite su uso, distribución y reproducción en cualquier medio, sin restricciones siempre que sin fines comerciales y que el trabajo original sea debidamente citado.
