










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.15 no.1 Madrid ene./mar. 2023 Epub 29-Mayo-2023
https://dx.doi.org/10.20960/revosteoporosmetabminer.000008
REVIEW
Genome-wide association studies (GWAS) vs functional validation: the challenge of the post-GWAS era
2Department of Genetics, Microbiology and Statistics. Faculty of Biology. Universitat de Barcelona. Barcelona, Spain
IBUB. Universitat de Barcelona. Barcelona
Over the past few years, efforts have been made to determine the variants and genes that may be important to determine bone mineral density (BMD) that, at the same time, are involved in several bone diseases. To achieve this, the approach that has been the most successful of all has been genome-wide association studies (GWAS). In particular, in research on bone biology over 50 different large GWAS or GWAS metanalyses have been published identifying a total of 500 genetic loci associated with different bone parameters such as BMD, bone resistance, and risk of fracture. Although the discovery of associated variants is an essential aspect, the functional validation of such variants is equally important to elucidate their effect, as well as the causal correlation they have with genetic disease. Since it is a much more time consuming and tedious aspect it has become the new challenge of this post-GWAS era. Among the genes that have already been studied several Wnt signaling pathway genes have been included, among them, the SOST gene that plays a crucial role both determining the BMD of the population and monogenic diseases with elevated bone mass giving rise to a new therapy against osteoporosis. In this review we’ll be collecting the main GWAS associated with bone phenotypes, as well as some functional validations undertaken to analyze the associations found in them.
Keywords: Genome-wide association studies; Functional validation; Bone mineral density; Bone diseases
GENOME-WIDE ASSOCIATION STUDIES (GWAS)
Over the past few years, genome-wide association studies (GWAS) have been an essential tool to identify what genes are involved in complex diseases (1). These studies consist of establishing an association between the genetic or allelic frequency of millions of SNP (single nucleotide polymorphisms) type markers distributed across the genome and a particular phenotype or disease (2). This approach is the most complete and impartial tool that exists for the particular of complex diseases. Unlike candidate gene association studies, GWAS are a hypothesis-free approximation hypothesis that allows the discovery of new genes or signaling pathways involved in a given phenotype that, up until now, were completely unknown (3). GWAS has been possible thanks to new advances made in high-throughput genome technology, study design, improved statistical analysis, and the possibility of having large biobanks available (4,5). Due to the large number of simultaneous statistical tests performed and, therefore, the statistical corrections made (that require a threshold p value of 5x10-8 to be considered statistically significant at whole genome level, and the small effect each variant presents in complex diseases, extremely large cohorts are required. This has been achieved through metanalyses of the GWAS where different studies have come together to increase the size of the sample (6,7).
Although with the evident success reported, GWAS have 3 main limitations. First, the genetic variants used to validate the association with the particular phenotype are SNP markers (tagSNPs) that are homogeneously distributed across the whole genome with a minor allele frequency (MAF) ≥ 5 % in the population. Therefore, rare variants with possible strong effects in the phenotype are not included in these studies. An attempt has been made to solve this limitation by including variants of less frequency in genotype chips, whole exome/genome sequencing, WES/WGS) and/or using the phenotypic extremes of the cohorts. Second, the success of GWAS largely depends on the size of the sample. Therefore, as commented above, the most widely used strategy today is to establish large consortia including different cohorts from across the world. Therefore, super-cohorts of greater statistical power —but genetically heterogeneous— are obtained in such a way that variants of a specific population are very difficult to find. Third, GWAS report the most statistically relevant SNP called lead SNP. Although this SNP can be the one causing this association, other variants that are in linkage disequilibrium with respect to the lead SNP variant can be responsible too. If the SNP associated is found in a codifying region and involves a change of amino acid, chances are that the SNP will be causal. However, truth is that most lead SNPs can be found in non-codifying regions (96 %) both intronic (41 %) and intergenic (54 %), which complicates the demonstration of their causal roles. Due to their non-coding nature, conducting functional studies of these lead SNPs is truly challenging (8-10). Therefore, these functional studies are still scarce to this date, and establishing the functional basis of the associations found in such analyses is still to be elucidated in this post-GWAS era.
To conduct functionality studies, interdisciplinary approaches are needed including in silico analyses (computational approaches) (11,12) —like pathogenicity prediction tools—, in vitro studies including, among other, studies of the reporter gene assays (eg, luciferase) (13) and in vivo studies of animal models like the zebra fish or mice (14,15).
This review summarizes the main GWAS published to this date using skeletal phenotypes, followed by in vitro and in vivo studies generated from the first large GWAS metanalysis (16) ever conducted on bone mineral density (BMD) and risks of fracture.
GWAS AND BONES
To conduct GWAS of bone diseases such as osteoporosis, parameters like BMD, and the geometry and microarchitecture of the bone can be taken into consideration. Among these properties, the most widely used and the one that best predicts osteoporotic fracture is BMD that is a quantitative trait measured in a continuous scale using methods like dual-energy X-ray absorptiometry (DXA). It is estimated that BMD is a trait with an approximate heritability between 50 % and 80 %. Similarly, the geometry of the bone shows heritability rates between 30 % and 70 % while bone microarchitecture determined by high-resolution peripheral quantitative computed tomography scan (HR-pQCT) shows heritability rates between 20 % and 80 % (17).
Up until now, over 50 large GWAS have been conducted using bone parameters together with a plethora of GWAS in smaller and more homogenous cohorts. With this over 500 associated loci have been identified. Although the percentage of variance explained through GWAS has increased substantially over the past few years thanks to the use of larger cohorts, all these loci only explain a small percentage (20 %) of genetic contribution to BMD (18,19). This has created a gap between the variability explained by genetic factors and BMD heritability probably due to overestimating heritability or the fact that other genetic factors like copy number variants (CNV) or epigenetics are not being taken into consideration (20).
All in all, GWAS have yielded significant findings like the association between the SOST and LRP5 genes —that had already been involved in monogenic skeletal disorders— and some skeletal phenotypes or the identification of new genes whose involvement in bone phenotypes was previously unknown (21). Table I shows some of the most relevant GWAS associated with BMD, most of which have been reported in the GWAS catalog (https://www.ebi.ac.uk/gwas/). To narrow it down, only studies with cohorts > 10 000 individuals have been considered.
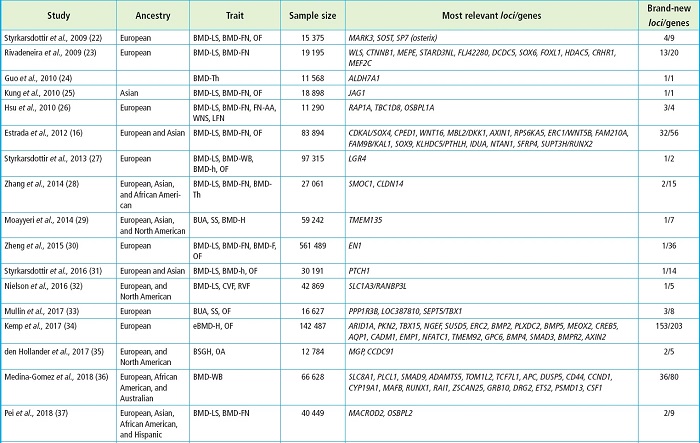
Table I (cont.). GWAS on bone and genes found with variants associated with skeletal phenotypes.
The study is represented by the first author and year. The genes are the study most relevant ones due to their association with skeletal phenotypes and their new finding. AA, axis angle; BLMAL, body lean mass of arms and legs; BMD, bone mineral density; BS, bone size; BSGH, bilateral semi-quantitative grading of the hand; BUA, broadband ultrasound attenuation; CVF, clinically confirmed vertebral fracture; DXA-h, X-ray absorptiometry of the shape of the hip; eBMD, estimated bone mineral density; F, forearm; FN, femoral neck; H, heel; h, hip; HU, heel ultrasound; LFN, length of the femoral neck; LS-BA, lumbar spine-bone area; LS, lumbar spine; MSFN, modular section of femoral neck; OA, osteoarthritis; OF, osteoporotic fracture; RVF, radiographically confirmed vertebral fracture; SS, speed of sound; Th, total hip; TLM, trunk lean mass; WB, whole body; WNS, width of the neck narrow section.
Many of the GWAS displayed on table I correspond to studies in which large metanalyses have been conducted leaving as a result hundreds of variants in different loci associated with skeletal phenotypes. However, most of these studies lack functional approaches.
FUNCTIONAL STUDIES IN THE POST-GWAS ERA
Despite the huge amount of association studies conducted to this date, functional studies have not developed at the same pace. Therefore, only a small fraction (164; 15 %) of the 1051 manuscripts that have cited the first large GWAS metanalysis on bone density (16) included functional studies whether in vitro or in vivo.
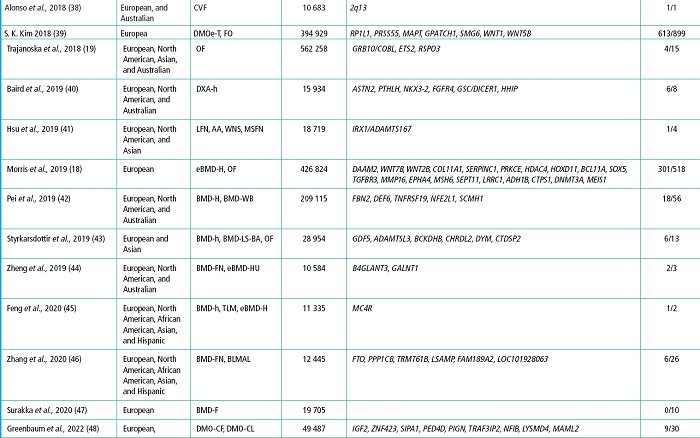
An example of successful functional studies is the characterization of the regulation of the SOST gene. This gene codes the sclerostin protein, a canonical Wnt signaling pathway inhibitor (49-51) associated with multiple bone parameters in different association studies across several populations (17,28,33,38,40,43,52,53) (Fig. 1A). Its inhibitory function on bone formation has been widely studied in in vivo and in vitro models. Currently, antisclerostin antibodies are used to treat bone diseases like osteoporosis or osteogenesis imperfecta (54-59). Therefore, the regulatory factors of the expression of the SOST gene are included among the new candidates as a target for the development of new therapies. In humans, SOST gene variants have been associated with conditions characterized by an excessive bone formation: sclerosteosis, craniodiaphyseal dysplasia, and the phenotypic trait of high bone mass (60) (Fig. 1B). To these diseases we may add Van Buchem disease. It is due to the deletion of the enhancer element ECR5 of SOST situated at the 52 kb region downstream of the gene that is necessary for the proper expression of the SOST gene (61) (Fig. 1A). Actually, the transcription of the SOST gene is finely regulated by many different signals both through direct regulation on the promoter of the SOST gene and through the distal ECR5 regulatory region (62,63) whose physical interaction has been demonstrated in a study recently conducted by our group on bone cells (64) (Fig. 1A). The MEF2C transcription factor is the best described SOST regulator in relation to its expression in osteocytes (63,65). The importance of MEF2C in the enhancer effect of ECR5 has been confirmed in the knock-out mouse model of Mef2c in osteoblasts/osteocytes that has a high bone mass and low levels of sclerostin (66). Precisely, MEF2C is yet another of the most repeated signals in GWAS with bone parameters (16,23,36,37,67-70). Together with MEF2C, HDAC5 has also been described as a negative regulator of the expression of the SOST gene that exerts its function by blocking the association of MEF2C and ECR5 during the differentiation of immature osteocytes (Fig. 1C). Consistent with this, the HDAC4/5 knock-out mouse model displays low BMD, and high expression of the SOST gene (71-73). Once again, HDAC4/5 is found among the most repeated loci in association studies with bone parameters (18,23,34,39,74) (Fig. 1B).
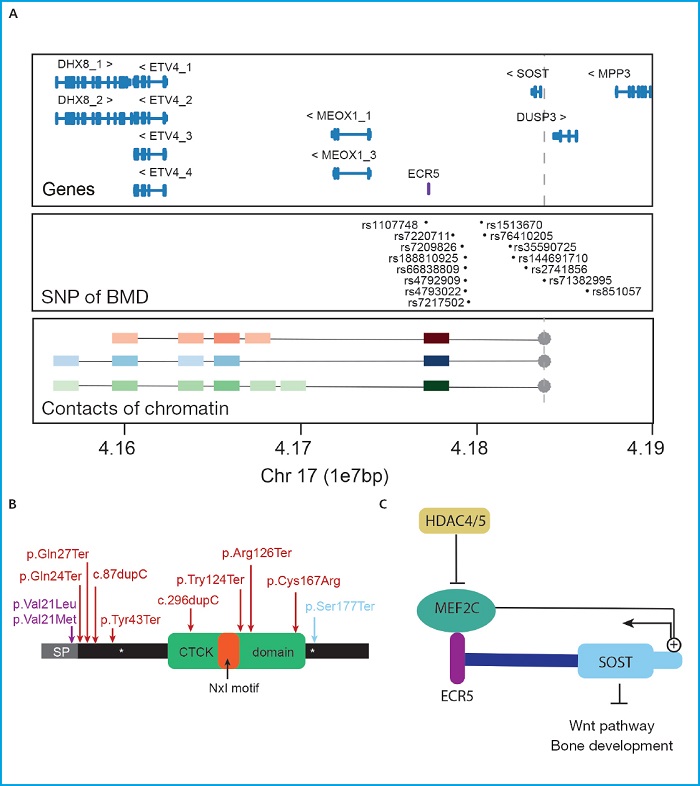
Figure 1. The SOST gene. A. Upper panel: Locus containing the SOST gene and its neighboring genes (GRC37/hg19). In purple, the ECR5 regulatory region. Main panel: SNPs associated with different bone parameters across different GWAS from the GWAS catalogue (https://www.ebi.ac.uk/gwas7). Lower panel: Main results of the 4C clinical trial conducted by Martínez-Gil et al. back in 2021 showing the main interactions of the SOST promoter (used as a bait and indicated with a dot and gray discontinuous). Colored squares show the interactions with color intensity proportional to the intensity of the interaction. Red, blue, and green squares show interactions with mesenchymal stem cells, hFOB cells, and SAOS2 cells, respectively. The units of the genomic scale used (1e7pb) correspond to 10 mega bases (1x107 base pairs). B. Schematic representation showing of sclerostin protein showing its functional domains and variants responsible for human skeletal conditions. Purple, red, and blue colors show the variants associated with craniodiaphyseal dysplasia, sclerosteosis, and the HBM phenotype variant. CTCK, C/terminal cysteine knot-like. C. Scheme of some of the positive and negative regulators of the expression of the SOST gene.
Another example of how important it is to conduct functional studies of associated regions is the DKK1 locus. This is another canonical Wnt signaling pathway inhibitor that plays a crucial role in the morphogenesis of the head (75,76), and bone development (77,78). Currently, no DKK1 variant has been described causing bone diseases in the HGMD database. Despite of this, our group identified 2 different missense variants in patients with the high BMD phenotype who show a functional loss of their inhibitory ability (13,79). On the other hand, one of these variants has also been found in patients with totally opposed phenotypes like osteoporosis or anal malformations (80,81). Also, we should mention that no GWAS has ever found SNPs in DKK1 associated with BMD or other bone parameters. However, an association with BMD has been demonstrated in a set of SNPs grouped in a region 350 kb downstream of DKK1 and 92 kb upstream of MBL2 (16,18,19,29,33,34,36,37,39,74) (Fig. 2). To distinguish which one of these 2 genes was responsible for this association, a study from our group (13) conducted a 4C chromatin conformation capture using the GWAS signal-rich region as a bait in 3 bone cellular types. This confirmed the physical interaction between this region and the DKK1 promoter ruling out any interaction with the MBL2 gene (Fig. 2; lower panel). It is precisely in this region where the LNCAROD gene is found, which specifies a DKK1 activator long noncoding (lncRNA), a possible culprit of the association found in the GWAS (82).
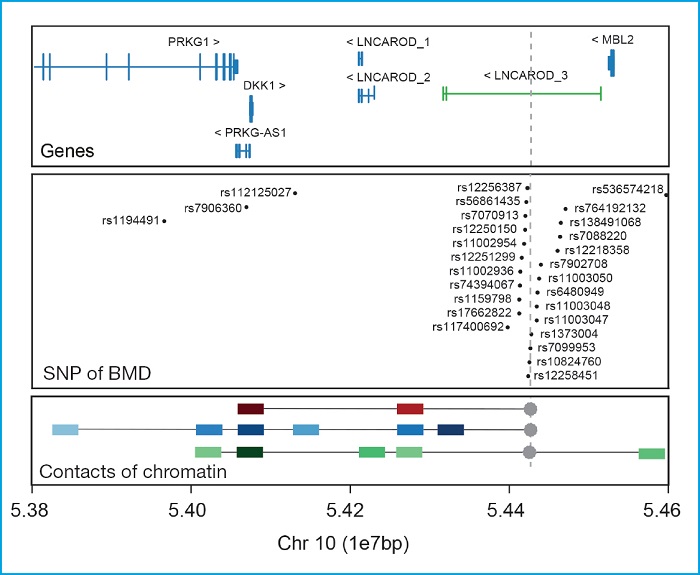
Figure 2. DKK1. Upper panel: locus containing the DKK1 gene and its neighboring genes (GRC37/hg19). In green, the lncRNA LNCAROD of GENCODE v32.2 (GRC38/hg18). Main panel: SNPs associated with different bone parameters across different GWAS taken from the GWAS catalogue (https://www.ebi.ac.uk/gwas7). Lower panel: Main results from the 4C clinical trial conducted by Martínez-Gil et al. in 2020 showing the main interactions with the SNP-rich region associated with BMD (used as a bait and indicated with a dot and gray discontinuous line). Colored squares show interactions with color intensity proportional to the intensity of the interaction. Red, blue, and green squares show interactions with mesenchymal stem cells, hFOB cells, and SAOS2 cells, respectively. The units of the genome scale used (1e7pb) correspond to10 mega bases (1x107 base pairs).
One of the most consistent loci across different GWAS on BMD is the genomic region situated in 7q31.31 including the WNT16 gene. This is a very complex loci, also including, apart from the WNT1 gene, the neighboring genes ING3, FAM3C, and CPED1. The role of the WNT16 gene determining BMD has been clearly established in functional studies of knock-out mouse models or osteoblast-specific conditional knock-out mice (6,83,84) that, largely, show spontaneous fractures due to low BMD plus reduced cortical thickness and bone resistance. However, evidence has been found on the importance of 3 other neighboring genes in bone metabolism. In the case of the protein coding gene ING3 (Inhibitor of Growth Family Member 3) —part of the Nucleosome Acetyltransferase of H4 histone acetylation (NuA4 HAT) complex involved in chromatin regulation— it has been found abundantly expressed in bone tissue (85).
In addition, functional studies of an in vitro cellular model of mesenchymal cells knocked-out for ING3 show osteoblastogenesis damage and stimulation of adipogenic differentiation (86). Regarding the CPED1 gene (Cadherin Like And PC-Esterase Domain Containing 1), no specific function of this gene has been found in humans or mice. However, in mice, functional studies show that the Cped1 gene is uniformly expressed in a variety of tissues including bone. Also, different isoforms have been described due to alternative splicing, as well as 3 promoter regions active during osteogenic differentiation (87). To better define its possible role in bone homeostasis, additional functional studies would be needed in in vitro cellular or animal models. FAM3C (family of sequence similarity 3c) is a cytokine-like growth factor expressed in multiple tissues (88) that plays a very important role in epithelial-mesenchymal transition, and cancer metastasis (89). Its association with bone metabolism has been confirmed with the knock-out mouse model that shows bone structure alterations (88).
Several functional studies have been conducted on the expression regulation of different genes at that region. For example, our group has conducted eQTL studies (expression Quantitative Trait Locus) with primary osteoblasts that show that SNPs located inside the WNT16 gene regulate the levels of expression of FAM3C of those cells (90). Also, in cells of osteoblastic lineage we have seen a physical interaction among different gene enhancers located inside the CPED1 gene, and the promoter of the WNT16 gene (91). All this shows the existence of a complex relation among these 4 genes, and suggests the possibility that they are working together. All in all, additional functional studies should be conducted to elucidate the role played by each of these genes, as well as all their possible interactions.
The aforementioned studies reveal the importance of functional studies based on the findings brought by analyzing GWAS. Challenge, now, is in the post-GWAS era. If we keep finding correlations between different variants in GWAS and functional aspects of these variants —in silico, in vitro or in vivo— we may end up finding new approaches and, therefore, new insights and therapeutic options for associated conditions and disorders.
REFERENCES
1. Lichou F, Gosia T. Functional studies of GWAS variants are gaining momentum. Nat Commun 2020;11(1):6283. DOI: 10.1038/s41467-020-20188-y [ Links ]
2. Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med 2010;363(2):166-76. DOI: 10.1056/NEJMra0905980 [ Links ]
3. Cannon ME, Karen LM. Deciphering the emerging complexities of molecular mechanisms at GWAS loci. Am J Hum Genet 2018;103(5):637-53. DOI: 10.1016/j.ajhg.2018.10.001 [ Links ]
4. Vissecher PV, Wray N, Zhang Q, Sklar P, McCarthy M, Brown M, et al. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101(1):5-22. DOI: 10.1016/j.ajhg.2017.06.005 [ Links ]
5. Buniello A, MacArthur J, Cerezo M, Harris L, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47(D1):D1005-12. DOI: 10.1093/nar/gky1120 [ Links ]
6. Medina-Gomez C, Kemp J, Estrada K, Eriksson J, Liu J, Reppe S, et al. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet 2012;8(7):e1002718. DOI: 10.1371/journal.pgen.1002718 [ Links ]
7. Loic Y, Sidorenko J, Kemper K, Zheng Z, Wood A, Weedon M, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet 2018;27(20):3641-9. DOI: 10.1093/hmg/ddy271 [ Links ]
8. Tam V, Patel N, Turcotte M, BosséY, ParéG, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet 2019;20(8):467-84. DOI: 10.1038/s41576-019-0127-1 [ Links ]
9. Hirschhorn JN. Genomewide association studies — illuminating biologic pathways. N Engl J Med 2009;360(17):1699-701. DOI: 10.1056/NEJMp0808934 [ Links ]
10. Klein RJ, Xing X, Mukherjee S, Willis J, Hayes J. Successes of genome-wide association studies. Cell 2010;142(3):350-1. [ Links ]
11. Schaid DJ, Chen W, Larson NB. From Genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 2018;19(8):491-504. [ Links ]
12. Broekema RV, Bakker OB, Jonkers IH. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol 2020;10(1):190221. DOI: 10.1098/rsob.190221 [ Links ]
13. Martínez-Gil N, Roca-Ayats N, Atalay N, Pineda-MoncusíM, Garcia-Giralt N, Van Hul W, et al. Functional assessment of coding and regulatory variants from the DKK1 locus. JBMR plus 2020;4(12):e10423. DOI: 10.1002/jbm4.10423 [ Links ]
14. Schartl M. Beyond the zebrafish: diverse fish species for modeling human disease. Dis Model Mech 2014;7(2):181-92. DOI: 10.1242/dmm.012245 [ Links ]
15. Rao S, Yao Y, Bauer DE. Editing GWAS: experimental approaches to dissect and exploit disease-associated genetic variation. Genome Med 2021;13(1):41. DOI: 10.1186/s13073-021-00857-3 [ Links ]
16. Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet 2012;44(5):491-501. DOI: 10.1038/ng.2249 [ Links ]
17. Trajanoska K, Rivadeneira F. The genetic architecture of osteoporosis and fracture risk. Bone 2019;126:2-10. [ Links ]
18. Morris JA, Kemp JA, Youlten SE, Laurent L, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet 2019;51(2):258-66. [ Links ]
19. Trajanoska K, Morris JA, Oei L, Zheng H-F, Evans DM, Kiel DP, et al. Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ 2018;362:k3225. DOI: 10.1016/j.bone.2019.04.005 [ Links ]
20. García-Ibarbia C, Delgado-Calle J, Casafont I, Velasco J, Arozamena J, Pérez-Núñez M, et al. Contribution of genetic and epigenetic mechanisms to Wnt pathway activity in prevalent skeletal disorders. Gene 2013;532(2):165-72. DOI: 10.1016/j.gene.2013.09.080 [ Links ]
21. Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, et al. LRP4 third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet 2014;23(7):1856-68. DOI: 10.1093/hmg/ddt578 [ Links ]
22. Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. New sequence variants associated with bone mineral density. Nat Genet 2009;41(1):15-7. DOI: 10.1038/ng.284 [ Links ]
23. Rivadeneira F, Styrkársdottir U, Estrada K, Halldórsson BV, Hsu Y-H, Richards JB, et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet 2009;41(11):1199-206. DOI: 10.1038/ng.446 [ Links ]
24. Guo Y, Tan L-J, Lei S-F, Yang T-L, Chen X-D, Zhang F, et al. Genome-wide association study identifies ALDH7A1 as a novel susceptibility gene for osteoporosis. PLoS Genet 2010;6(1):e1000806. DOI: 10.1371/journal.pgen.1000806 [ Links ]
25. Kung AW, Xiao SM, Cherny S, Li GH, Gao Y, Tso G, et al. Association of JAG1 with bone mineral density and osteoporotic fractures: a genome-wide association study and follow-up replication studies. Am J Hum Genet 2010;86(2):229-39. DOI: 10.1016/j.ajhg.2009.12.014 [ Links ]
26. Hsu YH, Zillikens MC, Wilson SG, Farber CR, Demissie S, Soranzo N, et al. An integration of genome-wide association study and gene expression profiling to prioritize the discovery of novel susceptibility loci for osteoporosis-related traits. PLoS Genet 2010;6(6):e1000977. DOI: 10.1371/journal.pgen.1000977 [ Links ]
27. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Aslaug J, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 2013;497(7450):517-20. DOI: 10.1038/nature12124 [ Links ]
28. Zhang L, Choi HJ, Estrada K, Leo PJ, Li J, Pei Y-F, et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum Mol Genet 2014;23(7):1923-33. DOI: 10.1093/hmg/ddt575 [ Links ]
29. Moayyeri A, Hsu Y-H, Karasik D, Estrada K, Xiao S-M, Nielson C, et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum Mol Genet 2014;23(11):3054-68. DOI: 10.1093/hmg/ddt675 [ Links ]
30. Zheng H-F, Forgetta V, Hsu Y-H, Estrada K, Rosello-Diez A, Leo PJ, et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 2015;526(7571):112-7. DOI: 10.1038/nature14878 [ Links ]
31. Styrkarsdottir U, Thorleifsson G, Gudjonsson SA, Sigurdsson A, Center JR, Hun Lee S, et al. Sequence variants in the PTCH1 gene associate with spine bone mineral density and osteoporotic fractures. Nat Commun 2016;7:10129. DOI: 10.1038/ncomms10129 [ Links ]
32. Nielson C, Liu C-T, Smith AV, Ackert-Bicknell C, Reppe S, Jakobsdottir J, et al. Novel genetic variants associated with increased vertebral volumetric BMD, reduced vertebral fracture risk, and increased expression of SLC1A3 and EPHB2. J Bone Miner Res 2016;31(12):2085-97. DOI: 10.1002/jbmr.2913 [ Links ]
33. Mullin BH, Zhao JH, Brown SJ, Perry J, Luan J, Zheng H-F, et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum Mol Genet 2017;26(14):2791-802. DOI: 10.1093/hmg/ddx174 [ Links ]
34. Kemp JP, Morris JA, Medina-Gomez C, Forgetta V, Warrington NM, Youlten SE, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet 2017;49(10):1468-75. DOI: 10.1038/ng.3949 [ Links ]
35. Hollander W, Boer CG, Hart DJ, Yau MS, Ramos Y, Metrustry S, et al. Genome-wide association and functional studies identify a role for matrix Gla protein in osteoarthritis of the hand. Ann Rheum Dis 2017;76(12):2046-53. DOI: 10.1136/annrheumdis-2017-211214 [ Links ]
36. Medina-Gomez C, Kemp JP, Trajanoska K, Luan J, Chesi A, Ahluwalia TS, et al. Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am J Hum Genet 2018;102(1):88-102. DOI: 10.1016/j.ajhg.2017.12.005 [ Links ]
37. Pei Y-F, Hu W-Z, Yan M-W, Li C-W, Liu L, Yang X-L, et al. Joint study of two genome-wide association meta-analyses identified 20p12.1 and 20q13.33 for bone mineral density. Bone 2018;110:378-85. DOI: 10.1016/j.bone.2018.02.027 [ Links ]
38. Alonso N, Estrada K, Albagha OME, Herrera L, Reppe S, Olstad OK, et al. Identification of a novel locus on chromosome 2q13, which predisposes to clinical vertebral fractures independently of bone density. Ann Rheum Dis 2018;77(3):378-85. DOI: 10.1136/annrheumdis-2017-212469 [ Links ]
39. Kim SK. Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PloS One 2018;13(7):e0200785. DOI: 10.1371/journal.pone.0200785 [ Links ]
40. Baird DA, Evans DS, Kamanu FK, Gregory JS, Saunders FR, Giuraniuc CV, et al. Identification of novel loci associated with hip shape: a meta-analysis of genome wide association studies. J Bone Miner Res 2019;34(2):241-51. DOI: 10.1002/jbmr.3605 [ Links ]
41. Hsu Y-H, Estrada K, Evangelou E, Ackert-Bicknell C, Akesson K, Beck T, et al. Meta-analysis of genomewide association studies reveals genetic variants for hip bone geometry. J Bone Miner Re. 2019;34(7):1284-96. DOI: 10.1002/jbmr.3698 [ Links ]
42. Pei YF, Liu L, Liu TL, Yang XL, Zhang H, Wei XT, et al. Joint association analysis identified 18 new loci for bone mineral density. J Bone Miner Re. 2019;34(6):1086-94. DOI: 10.1002/jbmr.3681 [ Links ]
43. Styrkarsdottir U, Stefansson OA, Gunnarsdottir K, Thorleifsson G, Lund SH, Stefansdottir L, et al. GWAS of bone size yields twelve loci that also affect height, BMD, osteoarthritis or fractures. Nat Commun 2019;10(1):2054. [ Links ]
44. Zheng J, Maerz W, Gergei I, Kleber M, Drechsler C, Wanner C, et al. Mendelian randomization analysis reveals a causal influence of circulating sclerostin levels on bone mineral density and fractures. J Bone Miner Res 2019;34(10):1824-36. DOI: 10.1038/s41467-019-09860-0 [ Links ]
45. Feng GJ, Wei XT, Zhang H, Yang XL, Shen H, Tian Q, et al. Identification of pleiotropic loci underlying hip bone mineral density and trunk lean mass. J Hum Genet 2020;66(3):251-60. DOI: 10.1038/s10038-020-00835-4 [ Links ]
46. Zhang YX, Zhang SS, Ran S, Liu Y, Zhang H, Yang XL, et al. Three pleiotropic loci associated with bone mineral density and lean body mass. Mol Genet Genomics 2020;296(1):55-65. DOI: 10.1007/s00438-020-01724-3 [ Links ]
47. Surakka I, Fritsche LG, Zhou W, Backman J, Kosmicki JA, Lu H, et al. MEPE loss-of-function variant associates with decreased bone mineral density and increased fracture risk. Nat Commun 2020;11(1):4093. DOI: 10.1038/s41467-020-17315-0 [ Links ]
48. Greenbaum J, Su KJ, Zhang X, Liu Y, Liu A, Zhao LJ, et al. A multiethnic whole genome sequencing study to identify novel loci for bone mineral density. Hum Mol Genet 2022;31(7):1067-81. DOI: 10.1093/hmg/ddab305 [ Links ]
49. Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem 2011;286(22):19489-500. DOI: 10.1074/jbc.M110.190330 [ Links ]
50. Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PloS One 2009;4(11):e7930. DOI: 10.1371/journal.pone.0007930 [ Links ]
51. Chang M-K, Kramer I, Huber T, Kinzel B, Guth-Gundel S, Leupin O, et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci USA 2014;111(48):E5187-195. DOI: 10.1073/pnas.1413828111 [ Links ]
52. Wang R, Zhao P, Kong N, Lu R, Pei Y, Huang C, et al. Genome-wide identification and characterization of the potato bHLH transcription factor family. Genes 2018;22;9(1):54. DOI: 10.3390/genes9010054 [ Links ]
53. Velázquez-Cruz R, Jiménez-Ortega RF, Parra-Torres AY, Castillejos-López M, Patiño N, Quiterio M, et al. Analysis of association of MEF2C, SOST and JAG1 genes with bone mineral density in Mexican-Mestizo postmenopausal women. BMC Musculoskelet Disord 2014;15:400. DOI: 10.1186/1471-2474-15-400 [ Links ]
54. Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med 2016;375(16):1532-43. DOI: 10.1056/NEJMoa1607948 [ Links ]
55. McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 2014;370(5):412-20. DOI: 10.1056/NEJMoa1305224 [ Links ]
56. Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res 2015;30(2):216-24. DOI: 10.1002/jbmr.2351 [ Links ]
57. Langdahl BL, Libanati C, Crittenden DB, Bolognese MA, Brown JP, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet 2017;390(10102):1585-94. DOI: 10.1016/S0140-6736(17)31613-6 [ Links ]
58. Glorieux FH, Devogelaer JP, Durigova M, Goemaere S, Hemsley S, Jakob F, et al. BPS804 anti-sclerostin antibody in adults with moderate osteogenesis imperfecta: results of a randomized phase 2a trial. J Bone Miner Res 2017;32(7):1496-504. DOI: 10.1002/jbmr.3143 [ Links ]
59. Lewiecki M, Blicharski T, Goemaere S, Lippuner K, Meisner PD, Miller PD, et al. A phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J Clin Endocrinol Metab 2018;103(9):3183-93. DOI: 10.1210/jc.2017-02163 [ Links ]
60. Martínez-Gil N, Ugartondo N, Grinberg D, Balcells S. Wnt pathway extracellular components and their essential roles in bone homeostasis. Genes (Basel) 2022;13(1):138. DOI: 10.3390/genes13010138 [ Links ]
61. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002;39(2):91-7. DOI: 10.1136/jmg.39.2.91 [ Links ]
62. Sebastian A, Loots GG. Genetics of Sost/SOST in sclerosteosis and van Buchem disease animal models. Metabolism 2018;80:38-47. DOI: 10.1016/j.metabol.2017.10.005 [ Links ]
63. Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, et al. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci USA 2012;109(35):14092-7. DOI: 10.1073/pnas.1207188109 [ Links ]
64. Martínez-Gil N, Roca-Ayats N, Cozar M, Garcia-Giralt N, Ovejero D, Nogués X, et al. Genetics and genomics of SOST: functional analysis of variants and genomic regulation in osteoblasts. Int J Mol Sci 2021;22(2):489. DOI: 10.3390/ijms22020489 [ Links ]
65. Loots G, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res 2005;15(7):928-35. DOI: 10.1101/gr.3437105 [ Links ]
66. Kramer I, Baertschi S, Halleux C, Keller H, Kneissel M. Mef2c deletion in osteocytes results in increased bone mass. J Bone Miner Res 2012;27(2):360-73. DOI: 10.1002/jbmr.1492 [ Links ]
67. Duncan E, Danoy P, Kemp JP, Leo PJ, McCloskey E, Nicholson GC, et al. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet 2011;7(4):e1001372. [ Links ]
68. Pei YF, Hu WZ, Hai R, Wang XY, Ran S, Lin Y, et al. Genome-wide association meta-analyses identified 1q43 and 2q32.2 for hip Ward's triangle areal bone mineral density. Bone 2016;91:1-10. DOI: 10.1016/j.bone.2016.07.004 [ Links ]
69. Zheng HF, Duncan EL, Yerges-Armstrong LM, Eriksson J, Bergström U, Leo PJ, et al. Meta-analysis of genome-wide studies identifies MEF2C SNPs associated with bone mineral density at forearm. J Med Genet 2013;50(7):473-8. DOI: 10.1136/jmedgenet-2012-101287 [ Links ]
70. Gregson CL, Newell F, Leo PJ, Clark GR, Paternoster L, Marshall M, et al. Genome-wide association study of extreme high bone mass: contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone 2018;114:62-71. DOI: 10.1016/j.bone.2018.06.001 [ Links ]
71. Baertschi S, Baur N, Lueders-Lefevre V, Voshol J, Keller H. Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation. J Biol Chem 2014;289(36):24995-5009. DOI: 10.1074/jbc.M114.564997 [ Links ]
72. Kobayashi Y, Uehara S, Koide M. Regulations of osteoclast formation and function by Wnt signals. Clin Calcium 2019;29(3):309-15. DOI: 10.20837/4201903309 [ Links ]
73. Wein M, Fretwurst T, Nahles S, Duttenhoefer F, Tomakidi P, Steinberg T, et al. Pilot investigation of the molecular discrimination of human osteoblasts from different bone entities. J Craniomaxillofac Surg 2015;43(8):1487-93. DOI: 10.1016/j.jcms.2015.07.030 [ Links ]
74. Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet 2019;104(1):65-75. [ Links ]
75. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci 2003;116(13):2627-34. DOI: 10.1016/j.jcms.2015.07.030 [ Links ]
76. Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998;391(6665):357-62. DOI: 10.1038/34848 [ Links ]
77. Ai M, Heeger S, Bartels CF, Schelling DK, Osteoporosis-Pseudoglioma Collaborative Group. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet 2005;77(5):741-53. DOI: 10.1086/497706 [ Links ]
78. Balemans W, Devogelaer JP, Cleiren E, Piters E, Caussin E, Van Hul W. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1-mediated inhibition of Wnt signaling. J Bone Miner Res 2007;22(5):708-16. DOI: 10.1359/jbmr.070211 [ Links ]
79. Martínez-Gil N, Roca-Ayats N, Monistrol-Mula A, García-Giralt N, Díez-Pérez A, Nogués X, et al. Common and rare variants of WNT16, DKK1 and SOST and their relationship with bone mineral density. Sci Rep 2018;8(1):10951. DOI: 10.1038/s41598-018-29242-8 [ Links ]
80. Korvala J, Löija M, Mäkitie O, Sochett E, Jüppner H, Schnabel D, et al. Rare variations in WNT3A and DKK1 may predispose carriers to primary osteoporosis. Eur J Med Genet 2012;55(10):515-9. DOI: 10.1016/j.ejmg.2012.06.011 [ Links ]
81. van de Putte R, Wijers CH, de Blaauw I, Feitz WF, Marcelis CL, Hakobjan M, et al. Sequencing of the DKK1 gene in patients with anorectal malformations and hypospadias. Eur J Pediatr 2015;174(5):583-7. DOI: 10.1007/s00431-014-2436-x [ Links ]
82. Ntini E, Louloupi A, Liz J, Muino JM, Marsico A, Vang-Ørom UA. Long ncRNA A-ROD activates its target gene DKK1 at its release from chromatin. Nat Commun 2018;9(1):1636. DOI: 10.1038/s41467-018-04100-3 [ Links ]
83. Zheng H-F, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet 2012;8(7):e1002745. DOI: 10.1371/journal.pgen.1002745 [ Links ]
84. Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med 2014;20(11):1279-88. DOI: 10.1038/nm.3654 [ Links ]
85. Nabbi A, Almami A, Thakur S, Suzuki K, Boland D, Bismar TA, et al. ING3 protein expression profiling in normal human tissues suggest its role in cellular growth and self-renewal. Eur J Cell Biol 2015;94(5):214-22. DOI: 10.1016/j.ejcb.2015.03.002 [ Links ]
86. Chesi A, Wagley Y, Johnson ME, Manduchi E, Su Ch, Lu S, et al. Genome-scale capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat Commun 2019;10(1):1260. DOI: 10.1038/s41467-019-09302-x [ Links ]
87. Maynard RD, Godfrey DA, Medina-Gomez C, Ackert-Bicknell CL. Characterization of expression and alternative splicing of the gene cadherin-like and PC esterase domain containing 1 (Cped1). Gene 2018;674:127-33. DOI: 10.1016/j.gene.2018.06.060 [ Links ]
88. Määttä JA, Bendre A, Laanti M, Büki KG, Rantakari P, Tervola P, et al. Fam3c modulates osteogenic cell differentiation and affects bone volume and cortical bone mineral density. Bonekey Rep 2016;5:787. DOI: 10.1038/bonekey.2016.14 [ Links ]
89. Bendre A, Büki KG, Määttä JA. Fam3c modulates osteogenic differentiation by down-regulating Runx2. Differentiation 2017;93:50-7. DOI: 10.1016/j.diff.2016.11.005 [ Links ]
90. Martínez-Gil N, Patiño J, Ugartondo N, Grinberg D, Balcells S. WNT16 rs2908004 missense variant acts as eQTL of FAM3C in human primary osteoblasts. Rev Osteoporos Metab Miner 2021;13(4):117-21. [ Links ]
91. Martínez-Gil N, Roca-Ayats N, Herrera C, Gritti N, Ugartondo N, Garcia-Giralt N, et al. Functional evidence of bone regulation of WNT16 through upstream enhancers within CPED1. J Bone Miner Res 2020;35S1:179. [ Links ]
Received: July 20, 2022; Accepted: December 22, 2022
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
