










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkArs Pharmaceutica (Internet)
On-line version ISSN 2340-9894
Ars Pharm vol.57 n.2 Granada Apr./Jun. 2016
https://dx.doi.org/10.30827/ars.v57i2.4962
ARTÍCULO ORIGINAL
Síntesis y actividad citotóxica de conjugados de la uridina con triterpenos en células de cáncer de mama
Synthesis and cytotoxic activity of conjugates of the uridine with triterpenes on breast cancer cells
Berrío Escobar Jhon Fernando1, Pastrana Restrepo Manuel Humberto1, Mejía Cuartas Diana Marcela1, Márquez Fernández Diana Margarita1, Márquez Fernández María Elena2 y Martínez Martínez Alejandro1
1 Grupo Productos Naturales Marinos, Facultad de Ciencias Farmacéuticas y Alimentarias, Universidad de Antioquia (UdeA), Calle 70 #5221, Medellín Colombia.
2 Universidad Nacional de Colombia, Sede Medellín Colombia, Facultad de Ciencias, Grupo Biotecnología Animal.
Los autores agradecen la financiación a la Universidad de Antioquia (Proyecto CODI CIQF-155 (2012-2014), Estrategia de Sostenibilidad 2014-2015, Beca Estudiante Instructor y programa Jóvenes Investigadores), a Colciencias (programa Jóvenes Investigadores).
Dirección para correspondencia
RESUMEN
Objetivos: Sintetizar conjugados del acetónido de la uridina con triterpenos (colesterol y 3β-5α,8α-endoperoxido-colest-6-en-3-ol) y ácido succínico como puente.
Métodos: Se preparó el acetónido de la uridina en acetona mediante catálisis ácida. Se prepararon los succinatos de los esteroles con anhídrido succínico y catalizador nucleofílico 4-N,N-dimetilamino-piridina (DMAP). Los conjugados 1 y 2 se sintetizaron mediante la esterificación de Steglich, con agente de acoplamiento N,N'-diciclohexilcarbodiimida (DCC)y DMAP. Los compuestos se caracterizaron por espectroscopia de RMN (1H RMN y 13C RMN) y espectrometría de masas. Los derivados se evaluaron sobre líneas celulares de ovario de hámster chino (CHO-K1) y de cáncer de mamá (MCF-7).
Resultados: Se obtuvieron derivados conjugados del acetónido de la uridina con dos triterpenos con rendimientos superiores al 80%. Los conjugados de uridina con triterpenos no presentaron inhibición significativa de la viabilidad celular sobre las líneas celulares MCF-7 y CHO-K1, tampoco se evidenció una relación dosis-respuesta para los compuestos evaluados.
Conclusiones: El método de esterificación con agentes de acoplamiento permitió obtener conjugados de la uridina con triterpenos empleando el ácido succínico como puente. Sin embargo los derivados de uridina obtenidos no presentaron actividad citotóxica significativa (p<0,05) sobre las líneas celulares evaluadas.
Palabras claves: actividad citotóxica, cáncer de mama, derivados de uridina, nucleósidos, triterpenos.
ABSTRACT
Aims: Synthesize of uridine acetonide conjugates with triterpenoids (cholesterol and 3β-5α,8α-endoperoxide-cholest-6-en-3-ol) and succinic acid as linking.
Methods: The acetonide derivative of uridine was prepared with acid catalysis in acetone. Sterols succinates were prepared with succinic anhydride and nucleophilic catalyst 4-N,N-dimethylamino-pyridine (DMAP). The conjugates were synthesized by Steglich method with N,N'-dicyclohexylcarbodiimide (DCC) Coupling agent and DMAP. The compounds were characterized by NMR spectroscopy (1H NMR, 13C NMR), and mass spectrometry. The derivatives were assessed in Chinese Hamster Ovary (CHO) and breast cancer (MCF-7) cell lines.
Results: The conjugates of uridine acetonide with two triterpenes were obtained with yields higher than 80%. The conjugates prepared don't showed significant inhibition of cell viability on MCF-7 and CHO cell lines, furthermore these substances did not show a relationship dose-response.
Conclusions: The esterification method with coupling agents allowed obtained uridine conjugates with triterpenoids. However the uridine derivatives don't showed significant cytotoxic activity (p<0,05) against cell lines evaluated.
Key words: breast cancer, cytotoxic activity, nucleosides, triterpenes, uridine derivatives.
Introducción
El cáncer de mama se encuentra entre los diez tipos más diagnosticados y de mayor mortalidad a nivel mundial1,2.
Los nucleósidos anticancerosos y análogos se emplean ampliamente desde la década de los 1960s, en la quimioterapia de tumores blandos y algunos tumores sólidos3,4. Estos fármacos presentan mecanismos de acción como anti-metabolitos: inhibición de la síntesis, transcripción, y desmetilación del DNA, desestabilización de la membrana mitocondrial e inducción y activación de rutas de apoptosis5,6. Sin embargo presentan desventajas farmacocinéticas (baja selectividad por las células cancerosas y gran labilidad química y biológica) y en la farmacodinamia (baja incorporación celular e inactivación intracelular por enzimas nucleósido desaminasas y/o ectonucleotidasas)7,8,9.
La uridina y algunos derivados fosfatados y glicosilados, son responsables de funciones celulares como moduladores en la formación de membranas y de la demanda energética, cofactores de glicosiltransferasas y agonistas de receptores purinérgicos (P2Xn y P2Ym)10,11. Algunos estudios encontraron que derivados glicosilados de la uridina, presentaron inhibición de enzimas glicosil-transferasas12. La uridina a concentraciones mayores a la fisiológica y derivados 3'-C-etiniluridina exhibieron potencial antitumoral sobre cáncer de mama y próstata10,11,13. Además se encontró que el tri-acetato de uridina presenta efectos moduladores sobre la toxicidad manifestada en la quimioterapia con fluorouracilo14.
Algunas investigadores aislaron 5α,8α-epidioxi-colesteroles de organismos marinos (ascidia Trididemnum sp. y erizo de mar Diadema sp.), los cuales presentaron actividad citotóxica significativa sobre líneas celulares de cáncer de mama MCF-7, sarcoma uterino FL, entre otros15,16.
Por lo expuesto anteriormente sobre la uridina y 5α,8α-epidioxi-colesteroles, se propuso la síntesis de conjugados de uridina con dos esteroles del colestano, vinculados mediante un di-ácido y la evaluación de su citotoxicidad en MCF-7.
Materiales y Métodos
Síntesis
Todos los solventes empleados fueron grado reactivo al igual los reactivos: Uridina, 99% (Alfaesar); anhídrido succínico, 99% (Merck), agente de acoplamiento N,N'-diciclohexilcarbodiimida (DCC), 99% y catalizador nucleofílico: 4-N,N-dimetilamino-piridina (DMAP), 99% (Alfaesar); colesterol, (99%) (Panreac) y 7,8-dehidro-colesterol, (99%) (Merck). Cromatografía en columna normal (sílica gel 60N) con gradiente: mezclas hexano/acetato de etilo y acetato de etilo/acetona. Los productos se analizaron por espectroscopia de resonancia magnética nuclear (1H-RMN a 300 MHz, 13C-RMN a 75 MHz, COSY H-H y HSQC), en un espectrómetro Bruker NMR 300 (300 MHz, Software Bruker TopSpin 3.1), solventes: cloroformo deuterado (CDCl3) (compuestos 1 y 2) y DMSO-d6 (acetónido y su succinato), ambos 99% pureza de deuterio (Merck), se empleó tetrametilsilano como estándar interno. También se analizaron en un espectrómetro de masa Agilent serie 6300 (ESI-Ion-Trap) en modo positivo por inyección directa.
3',4'-Acetónido de uridina o D3
Se partió de 500 mg de uridina, se adicionaron 30 μL del ácido inorgánico, se dejó bajo agitación constante por 3 horas17 (figura 1-a). Sólido amarillo, rendimiento 90%. NMR (300 MHz, DMSO-d6), δ ppm (multiplicidad; integral J (Hz); grupo; posición): 1,26 (s; 3H; -CH3; 3'b-H acetónido), 1,48 (s; 3H; -CH3; 3'c-H acetónido), 3,57 (m; 2H;-CH2O-; 6'-H ribosa (Rib)), 4,06 (q; 1H; J=4,2; -CH-; 5'-H Rib), 4,74 (dd; 1H; J=6,3 y 3,5; -CH-; 4'-H Rib), 4,89 (dd; 1H; J=6,3 y 2,6; -CH-; 3'-H Rib), 5,63 (d; 1H; J=8,0; -CH=; 5-H uracilo (U)), 5,82 (d; 1H; J=2,6; -CH-; 2'-H Rib), 7,79 (d; 1H; J=8,1; -CH=; 6-H U). 25,57 (1C; -CH3; 3'b-C acetónido), 27,49 (1C; -CH3; 3'c-C acetónido), 61,67 (1C; -CH2O-; 6'-C Rib), 79,61 (1C; -CH-; 4'-C Rib), 80,92 (1C; -CH-; 3'-C Rib), 84,13 (1C; -CH-; 5'-C Rib), 91,58 (1C; -CH-; 2'-C Rib), 102,19 (1C; -CH=; 5-C U), 113,43 (1C; -OCO-; 3'a-C acetónido), 142,47 (1C; -CH=; 6-C U), 150,79 (1C; N-CO-N; 2-C U), 163,67 (1C; -CO-N; 4-C U). MS (ESI-Ion-Trap) (m/z): Calculada para C12H16N2O6: 284,2652; encontrada 307,10 [M+Na]+.
Succinato del acetónido de la uridina o D4
Todas las reacciones de esterificación se hicieron con el método de Steglich. Se partió de 300 mg del acetónido de uridina, se adicionaron 0,5 equivalentes de anhídrido succínico y 5% molar de DMAP respecto al anhídrido, se dejó bajo agitación constante por 3 días (figura 1-a)18,19,20. Solido amarillo, rendimiento 85%. NMR (300 MHz, DMSO-d6), δ ppm (multiplicidad; integral J (Hz); grupo; posición): 1,26 (s; 3H; -CH3; 3'b-H acetónido), 1,48 (s; 3H; -CH3; 2'c-H acetónido), 2,60 (m; 4H;-CH2-; 6'b-H y 6'c-H ácido succínico (AS)) 4,40 (m; 3H;-CH2O- y -CH-; 6'-H y 5'-H ribosa (Rib)), 4,78 (m; 1H; -CH-; 4'-H Rib), 4,93 (m; 1H; -CH-; 3'-H Rib), 5,62 (d; 1H; J=8,0; -CH=; 5-H uracilo (U)), 5,81 (d; 1H; J=2,6; -CH-; 2'-H Rib), 7,80 (d; 1H; J=8,1; -CH=; 6-H U). 25,57 (1C; -CH3; 3'b-C acetónido), 27,49 (1C; -CH3; 3'c-C acetónido), 28,07 (2C; -CH2-; 6'b-C y 6'c-C AS), 61,74 (1C; -CH2O-; 6'-C Rib), 79,72 (1C; -CH-; 4'-C Rib), 81,02 (1C; -CH-; 3'-C Rib), 84,15 (1C; -CH-; 5'-C Rib), 91,08 (1C; -CH-; 2'-C Rib), 103,29 (1C; -CH=; 5-C U), 144,44 (1C; -CH=; 6-C U), 151,26 (1C; N-CO-N; 2-C U), 164,01 (1C; -CO-N; 4-C U), 171,05 (2C; -COOR; 6'a-C AS), 171,95 (1C; -COOR; 6'd-C AS). MS (ESI-Ion-Trap) (m/z): Calculada para C16H10N2O9: 384,338; encontrada 407,30 [M+Na]+.
Succinato de mono-colesterilo y succinato del 3β-5α,8α-endoperoxido-colest-6-en-3-ol (5α,8α-epidioxi-colesterol).
Se partió de 200 mg del esterol, y 5% molar de DMAP respecto al anhídrido (figura 1-b), se dejó bajo agitación por 4 días (figura 1-b)18,19,20. Para el succinato del 3β-5α,8α-endoperoxido-colest-6-en-3-ol (5α,8α-epidioxi-colesterol), se preparó previamente el epóxido a partir de 200 mg de 7,8-dehidrocolesterol en metanol, se añadió eosina (foto-sensibilizador), se sometió a flujo constante de aire y radiación con lámpara halógena, se dejó bajo agitación constante a temperatura ambiente por 24 horas21.
Síntesis de derivados conjugados de uridina-succinato-tri-terpeno
La preparación de los derivados conjugados del acetónido de la uridina con colesterol y 5,8-epidioxi-esterol, se realizó por dos rutas18,19,20.
a. Se partió de 150 mg del succínico mono-esterificado con acetónido de uridina, se adicionaron 1,2 equivalentes del esterol, luego 1,5 equivalentes de DCC y 5% molar de DMAP respecto al succinato (figura 1-a)
b. Se partió 150 mg del succínico mono-esterificado con esterol, se adicionaron 1,2 equivalentes del acetónido de uridina, el DCC y DMAP se adicionaron las mismas proporciones que en ruta (a) (figura 1-b).
3',4'-acetónido-uridina-succinato-3"β-5"α,8"α-endoperoxido-colest-6"-en-3"-ol conjugado (3',4'-acetónido-uridina-succinato-5"α,8"α-epidioxi-colesterol conjugado) o D1
Sólido blanco, rendimiento 90%. P.F. 93-94oC. NMR (300 MHz, CDCl3), δ ppm (multiplicidad; integral J (Hz); grupo; posición): 1,34 (s; 3H; -CH3; 3'b-H acetónido), 1,52 (s; 3H; -CH3; 2'c-H acetónido), 2,60 (m; 4H;-CH2-; 6'b-H y 6'c-H ácido succínico (AS)) 4,34 (m; 3H;-CH2O- y -CH-; 6'-H y 5'-H ribosa (Rib)), 4,80 (m; 1H; -CH-; 4'-H Rib), 4,97 (m; 2H; -CH-O; 3'-H Rib y 3"-H epidioxicolesterol (EDOColes)), 5,66 (d; 1H; J=2,6; -CH-; 2-H' Rib), 5,71 (d; 1H; J=8,1; -CH=; 5-H uracilo (U)), 6,26 (d; 1H; J=8,4; -CH=; 7"-H EDOColes), 6,55 (d; 1H; J=8,3; -CH=; 6"-H EDOColes), 7,31 (d; 1H; J=8,2; -CH=; 6-H U). 12,68 (1C; -CH3; 18"-C; EDOColes), 18,09 (2C; -CH3; 19"-C y 21"-C EDOColes), 22,59 (2C; -CH3; 26"-C y 27"-C EDOColes), 24,80 (1C; -CH3; 3'b-C acetónido), 25,76 (1C; -CH3; 3'c-C acetónido), 28,07 (2C; -CH2-; 6'b-C y 6'c-C AS), 63,06 (1C; -CH2O-; 6'-C Rib), 70,21 (1C; -CH-O; 3"-C EDOColes), 79,41 (1C; -CH-; 5"-C EDOColes), 81,17 (1C; -CH-; 4'-C Rib), 84,32 (1C; -CH-; 3'-C Rib), 84,88 (1C; -CH-; 5'-C Rib), 81,57 (1C; -CH-; 8"-C EDOColes), 94,55 (1C; -CH-; 2'-C Rib), 102,40 (1C; -CH=; 5-C U), 114,61 (1C; 3'a-C acetónido), 130,98 (1C; -CH=; 6"-C EDOColes), 134,94 (1C; -CH=; 7"-C EDOColes), 142,03 (1C; -CH=; 6-C U), 149,52 (1C; N-CO-N; 2-C U), 162,64 (1C; -CO-N; 4-C U), 171,09 (2C; -COOR; 6'a-C AS), 171,78 (1C; -COOR; 6'd-C AS). MS (ESI-Ion-Trap) (m/z): Calculada para C43H62N2O11: 782,9592; encontrada 805,60 [M+Na]+.
3',4'-acetónido-uridina-succinato-3"β-colest-6"-en-3"-ol (3',4'-acetónido-uridina-succinato-colesterol conjugado) o D2
Sólido pardo, rendimiento 80%. P.F. 72-73 oC. NMR (300 MHz, CDCl3), δ ppm (multiplicidad; integral J (Hz); grupo; posición): 1,25 (s; 3H; -CH3; 3'b-H acetónido), 1,48 (s; 3H; -CH3; 2'c-H acetónido), 2,35 (m; 1H; -CH-; 7"-H colesterol (Coles)), 2,64 (m; 4H; -CH2-; 6'b-H y 6'c-H ácido succínico (AS)) 4,40 (m; 3H; -CH2O- y -CH-; 6'-H y 5'-H ribosa (R)), 4,67 (m; 1H; -CH-O; 3"-H Coles), 4,82 (m; 1H; -CH-; 4'-H Rib), 5,05 (m; 1H; -CH-; 3'-H Rib), 5,48 (m; 1H; -CH=; 6"-H Coles), 5,75 (d; 1H; J=2,6; -CH-; 2-H' Rib), 5,80 (d; 1H; J=8,0; -CH=; 5-H uracilo (U)), 7,45 (d; 1H; J=8,1; -CH=; 6-H U). 12,66 (1C; -CH3; 18"-C Coles), 18,75 (2C; -CH3; 19"-C y 21"-C Coles), 22,43 (2C; -CH3; 26"-C y 27"-C Coles), 24,86 (1C; -CH3; 3'b-C acetónido), 25,47 (1C; -CH3; 3'c-C acetónido), 28,27 (2C; -CH2-; 6'b-C y 6'c-C AS), 32,27 (2C; -CH2- y -CH-, 7"-C y 8"-C Coles), 64,32 (1C; -CH2O-; 6'-C Rib), 74,98 (1C; -CH-O; 3"-C Coles), 81,79 (1C; -CH-; 4'-C Rib), 84,93 (1C; -CH-; 3'-C Rib), 85,11 (1C; -CH-; 5'-C Rib), 94,78 (1C; -CH-; 2'-C Rib), 102,88 (1C; -CH=; 5-C U), 114,84 (1C; 3'a-C acetónido), 123,50 (1C; -CH=; 6"-C Coles), 139,05 (1C; -CH; 5"-C Coles), 142,31 (1C; -CH=; 6-C U), 149,79 (1C; N-CO-N; 2-C U), 163,48 (1C; -CO-N; 4-C U), 171,96 (2C; -COOR; 6'a-C AS), 172,02 (1C; -COOR; 6'd-C AS). MS (ESI-Ion-Trap) (m/z): Calculada para C43H64N2O9: 752,9762; encontrada 775,60 [M+Na]+.
Efecto sobre la viabilidad celular
Líneas celulares CHO-K1 (ovario de hámster chino, ATCC No CCL61) y MCF-7 (adenocarcinoma de mama humano, ATCC No HTB22), medios de cultivo HAM F12 y DMEM (Sigma), suero bovino fetal (SBF, Gibco), penicilina100 U/mL-estreptomicina 100 µg/mL (Gibco) y bromuro de 3-(4,5-dimetiltiazol-2-ilo)-2,5-difeniltetrazol (MTT, Sigma). Solventes: DMSO grado reactivo, pureza 99,9% (Sigma) y 2-propanol grado reactivo, pureza 98% (J. T. Baker). Las absorbancias se leyeron en espectrofotómetro Multiskan FC Microplate Photometer-ThermoScientific.
Cultivo celular
Los experimentos se realizaron con cultivos de células CHO-K1 y MCF-7 en fase exponencial, en medio DMEM suplementado con 5% de SBF, penicilina 100 U/mL-estreptomicina 100 µg/mL, se incubaron a 37oC por 48 h. La línea celular MCF-7 se incubó en atmósfera húmeda con 5% de CO2.
Ensayo MTT
Las células MCF-7 y CHO-K1 fueron sembradas en medio DMEM o F-12 suplementado con SBF, en platos de 96 pozos con densidad celular de 5x103 células/pozo y 6x103 de células/pozo, respectivamente. Los cultivos se trataron con concentraciones de 1 μM, 10 μM y 100 μM de la uridina (Urd) o sus derivados, durante 48 horas a 37oC. Después se adicionó 10 μL de MTT (5 mg/mL) y se incubaron en la oscuridad a 37oC por 4 horas, luego a cada pozo se adicionó 100 μL de iso-propanol acidulado, se agitó por 1 hora y se midió la absorbancia a 570 nm. Se calculó el porcentaje de viabilidad celular de experimentos por duplicado de tres réplicas por cada tratamiento. Los resultados se expresaron como la media y la desviación estándar (X±SD) y el análisis estadístico se realizó con ANOVA de dos vías, el Test de Bonferroni con p<0.05 mediante el Software GraphPad Prism Versión 5.
Resultados
Aspectos sintéticos
El método de Steglich emplea agentes de acoplamiento y/o catalizadores nucleofílicos para activar los ácidos orgánicos en solventes orgánicos de mediana polaridad a temperatura ambiente, por lo tanto es un método ideal para trabajar con sustratos lábiles como la uridina, este permitió la obtención de los conjugados de uridina sin degradación alguna, con porcentajes de rendimiento del 80% y 90% para los conjugados con colesterol (2) y 5,8-epidioxicolesterol (1), respectivamente. Sin embargo los conjugados se obtuvieron solo mediante la secuencia (b) presentada en la metodología (figura 1-b).
Caraterización
Los conjugados de uridina con los triterpenos se identificaron por comparación con los datos espectroscópicos 1H-RMN a 300 MHz, 13C-RMN a 75 MHz, COSY H-H y HSQC de la uridina y el acetónido de la uridina. Los resultados de RMN y espectrometría de masas de los derivados conjugados de la uridina están de acuerdo con la estructura propuesta.
Actividad biológica
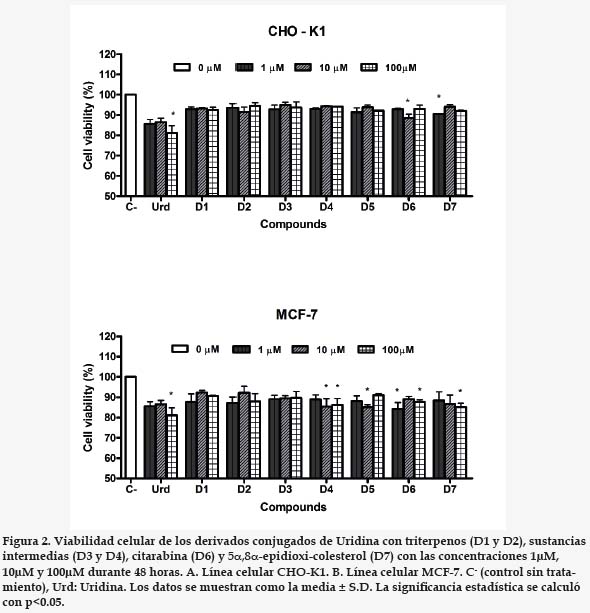
Los conjugados del acetónido de la uridina con triterpenos (D1 y D2), el acetónido de la uridina (D3), el succinato del acetónido (D4) y el epidioxicolesterol (D7) no presentaron inhibición significativa de la viabilidad celular sobre células CHO-K1 y MCF-7, aún bajo la concentración más alta evaluada (100 μM) (figura 2). La uridina (Urd) presentó citotoxicidad apreciable (alrededor del 20%) a la mayor concentración evaluada. Cabe destacar que la evaluación de la citarabina (D6) sobre las mismas líneas celulares presento resultados similares que la uridina.
Discusión
Aspectos sintéticos
Al ensayar dos secuencias para la obtención de los derivados conjugados de la uridina, se encontró que la ruta (b) arrojo los mejores resultados, esto se debió posiblemente a que el intermediario O-acil-isourea-b formado por el succinato del triterpeno es menos voluminoso y más reactivo ante un ataque nucleofílico que el derivado O-acil-isourea-a formado por el succinato del acetónido de uridina, el cual presenta grandes repulsiones estéricas entre los dos ciclohexilos, el uracilo y el acetónido cíclico, esto puede impedir el ataque del catalizador nucleofílico.
El acetónido de uridina presenta mayor solubilidad en solventes de polaridad media que el nucleósido, por lo tanto la probabilidad de choques efectivos y su acción como nucleófilo sobre el carbonilo de la amida cuaternaria (DMAP-succínico-esterol) se incrementa. Otra ventaja, se debe a que esta amida cuaternaria presenta una apreciable parte plana cerca al carbonilo, lo cual permite o facilita el ataque como nucleófilo del hidroxilo de la posición 6' de la ribosa, el cual se ubica anti al acetónido cíclico, minimizando así las repulsiones estéricas al momento del ataque.
Caracterización
Espectro de acoplamiento protón-protón (COSY H-H)
En el espectro para el compuesto 2, en campo alto, se observan los acoplamientos característicos de las cadenas y anillos del colesterol, los correspondientes al ácido succínico y a los metilos del acetónido. En campo medio, desplazamientos químicos entre (3,0-6,0) ppm, se observa que la señal multiplete en δ=4,40 ppm, (protones 6'-H (-CH2-O) y 5'-H (-CH-O) ribosa) esta acoplada con un multiplete más desprotegido δ=4,82 (protón 4'-H ribosa), este protón a su vez presenta acoplamiento con una señal de mayor desplazamiento δ=5,05 (protón 3'-H ribosa), la cual esta acoplada con una señal doblete más desprotegida δ=5,75 (protón 2'-H ribosa). Se observa que el multiplete en δ=5,48 (protón posición 6"-H colesterol) presenta varios acoplamientos con protones localizados en campo alto. En campo bajo (desplazamiento químico superior a 6 ppm), se observa que el doblete con el mayor desplazamiento δ=7,45 (protón 6-H uracilo) esta acoplado con un doblete más protegido δ=5.80 (protón 5-H uracilo). De igual forma se hizo el análisis para el compuesto 1.
Espectro de acoplamiento protón-carbono (HSQC)
En el espectro para el compuesto 2, en campo alto, se observan los acoplamientos característicos de las cadenas y anillos del colesterol, además de los correspondientes al ácido succínico, empleado como puente y a los metilos del acetónido. En campo medio, con desplazamientos de protón (3,0-6,0) ppm y carbono (50-110) ppm, se observa que la señal de los protones 6'-H y 5'-H de la ribosa, esta acoplada con dos clases de carbono, un carbono más protegido localizado δ=64,32 ppm (posición 6'-C ribosa) y otro carbono con mayor desplazamiento δ=85,11 ppm (posición 5'-C ribosa). Luego se ve que la señal del protón 4'-H ribosa esta acoplado con un carbono más protegido δ=81,79 ppm (posición 4'-C ribosa) y la señal del protón 3'-H ribosa esta acoplada con un carbono con mayor desprotección δ=84,93 ppm (posición 3'-C ribosa). Se ve posteriormente un acoplamiento entre la señal del protón 2'-H ribosa y un carbono que presenta mayor desplazamiento δ=94,78 ppm (carbono 2'-C ribosa). También se observa que la señal del protón olefinico del colesterol (posición 6"-H) esta acoplado con un carbono con gran desprotección δ=123,50 ppm (posición 6"-C colesterol).
En campo bajo, se observa que la señal del protón 5-H uracilo esta acoplada con un carbono vinílico con gran desprotección δ=102,88 ppm (posición 5-C uracilo). Por último se ve un acoplamiento entre la señal del protón 6-H uracilo con otro carbono vinílico con una mayor desprotección δ=142,31 ppm (posición 6-C uracilo). De igual forma se hizo el análisis para el compuesto 1.
Actividad biológica
Los resultados obtenidos para la uridina y citarabina (figura 2), sugieren una posible resistencia por la línea celular MCF-7 ante los nucleósidos, por tanto es de esperarse que los compuestos derivados de uridina no exhiban citotoxicidad significativa, la resistencia puede deberse a tres factores: A. la sobreproducción de enzimas blanco, como pyr1,3 y pyr5,6 que intervienen en la síntesis de novo de nucleósidos pirimidínicos, inhibiendo la ruta de salvamento de los mismos22.
B. la sobre-expresión de las fosfo-glicoproteinas trans-membranales, esto puede disminuir el transporte de nucleósidos del medio exterior a través de la membrana23.
C. La sobreexpresión de los receptores de tirosina quinasa (ErbBX), que modulan el ciclo celular, evitando la detención del ciclo celular inducido por los nucleósidos2.
Desde el punto de vista químico, la estructura de los conjugados 1 y 2 puede afectar la actividad, debido a que los triterpenos y el acetónido cíclico presentan un gran tamaño y orientación espacial, exhibirán grandes repulsiones estéricas que pueden afectar las interacciones con el sitio activo de las esterasas, quedando el carbonilo lejos o impedido para la reacción de hidrolisis o restitución del hidroxilo en la posición 6' de la ribosa, esencial para la activación de los nucleósidos (fosfatación por parte de las quinasas)9,19,24.
Conclusiones
El método de esterificación de Steglich, permitió obtener conjugados del acetónido de la uridina con triterpenos con rendimientos altos, sin embargo fue necesario utilizar un di-ácido como puente. La ruta de acoplamiento directo entre el succinato del acetónido y los esteroles no dio los resultados esperados. El acoplamiento entre los succinatos de los triterpenos con el acetónido se obtuvo con rendimientos altos, superiores al 80% y tiempos de reacción menores.
La uridina, los conjugados con triterpenos y las sustancias intermedias no presentan un efecto citotóxico significativo con p<0,05 para, tampoco se observa relación entre la citotoxicidad y la concentración de los mismos. La poca actividad exhibida por la uridina y citarabina, sugiere que las líneas celulares empleadas, particularmente la MCF-7, no son afectadas por los nucleósidos y pueden presentar algún mecanismo de resistencia.
Agradecimientos
Los autores agradecen la colaboración de la Joven Investigadora-Colciencias: Suly S. Villa V. y Jóvenes Investigadores-CODI: Angélica M. Bustamante y Álvaro J. García O.
Conflicto de interés
Los autores declaran que no existe conflicto de interés alguno en este trabajo.
Referencias
1. Chik F, Machnes Z, Szyf M. Synergistic anti-breast cancer effect of a combined treatment with the methyl donor s-adenosyl methionine and the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Carcinogenesis. 2014;35(1):138-144. [ Links ]
2. Strasser S, Maier S, Leisser C, et al. 5-fdurd-AraC heterodinucleoside re-establishes sensitivity in 5-fdurd- and AraC-resistant MCF-7 breast cancer cells overexpressing erbb2. Differentiation. 2006;74(9-10):488-498. [ Links ]
3. Silvestris N, Cinieri S, La Torre I, et al. Role of gemcitabine in metastatic breast cancer patients: A short review. Breast. 2008;17(3):220-226. [ Links ]
4. Mehta DR, Foon KA, Redner RL, et al. Fludarabine and cytarabine in patients with acute myeloid leukemia refractory to two different courses of front-line chemotherapy. Leuk Res. 2011;35(7):885-888. [ Links ]
5. Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phophorylases: properties, functions and clinical aspects. Pharmacol Ther. 2000;88(3):349-425. [ Links ]
6. Zhenchuk A, Lotfi K, Juliusson G, Albertioni F. Mechanisms of anti-cancer action and pharmacology of clofarabine. Biochem Pharmacol. 2009;78(11):1351-1359. [ Links ]
7. Wagner CR, Iyer VV, Mcintee EJ. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med Rev Res. 2000;20(6):417-451. [ Links ]
8. Sarpietro MG, Ottimo S, Giuffrida MC, Rocco F, Ceruti M, Castelli F. Synthesis of n-squalenoyl cytarabine and evaluation of its affinity with phospholipid bilayers and monolayers. Int J Pharm. 2011;406(1-2):69-77. [ Links ]
9. Moysan E, Bastiat G, Benoit J. Gemcitabine versus modified gemcitabine: a review of several promising chemical modification. Molecular pharmaceutics. 2013; 10:430-444. [ Links ]
10. Yamamoto T, Koyama H, Kurajoh M, Shoji T, Tsutsumi Z, Moriwaki Y. Biochemistry of uridine in plasma. Clin Chim Acta. 2011;412(19-20):1712-1724. [ Links ]
11. Lazarowski Er, Schwarzbaum Pj. Señales purinérgicas. Medicina. 2009;69(2):267-276. [ Links ]
12. Wandzik I, Bieg T, Czaplicka M. Synthesis of 2-deoxy-hexopyranosyl derivatives of uridine as donor substrate analogues for glycosyltransferases. Bioorg Chem. 2009;37(6):211-216. [ Links ]
13. Hrdlicka P.J, Jepsen J.S, Nielsen C, Wengela J. Synthesis and biological evaluation of nucleobase-modified analogs of the anticancer compounds 3'-C-ethynyluridine (Eurd) And 3'-C-ethynylcytidine (Ecyd). Bioorg. Med. Chem. 2005;13:1249-1260. [ Links ]
14. McEvillyn M, Popelas C, Tremmel B. Use of uridine triacetate for the management of fluorouracil overdose. Am J Health-Syst Pharm. 2011;68:1806-1809. [ Links ]
15. Ioannou E, Abdel-Razik AF, Zervou M, et al. 5α,8α-Epidioxysterols from the gorgonian Eunicella cavolini and the ascidian Trididemnum inarmatum: Isolation and evaluation of their antiproliferative activity. Steroids. 2009;74:73-80. [ Links ]
16. Minn C, Kiem P, Huong L, Kim Y. Cytotoxic constituents of Diadema setosum. Arch. Pharm. Res. 2004;27:734-7. [ Links ]
17. Clayden J, Greeves N, Warren S. and Wothers P. Organic Chemistry. 2dn Ed. Oxford: Oxford University Press. 2012. [ Links ]
18. Li W, Wu J, Zhan P, et al. Synthesis, drug release and anti-HIV activity of a series of PEGylated zidovudine conjugates. Int J Biol Macromol. 2012;50(4):974-980. [ Links ]
19. Radi M, Adema AD, Daft JR, et al. In Vitro Optimization of Non-Small Cell Lung Cancer Activity with Troxacitabine. J. Med. Chem. 2007;50(7):2249-2253. [ Links ]
20. Neises B, Steglich W. Simple method for the esterification for carboxylic acids. Angew Chem Int. 1978;17(7):522-524. [ Links ]
21. Valente P, Avery TD, Taylor DK, Tiekink ERT. Synthesis and Chemistry of 2,3-Dioxabicyclo(2.2.2)octane-5,6-diols. J. Org. Chem. 2009;74(1):274-282. [ Links ]
22. Karle JM, Cowan KH, Chisena CA, Cysyk RL. Uracil Nucleotide Synthesis in a Human Breast Cancer Cell Line (MCF-7) and in Two Drug-Resistant Sublines that Contain Increased Levels of Enzymes of the de Novo Pyrimidine Pathway. Molecular Pharmacology. 1986;30:136-1341. [ Links ]
23. Morgan PE, Fine RL, Montgomery P, Marangos PJ. Multidrug resistance in MCF-7 human breast cancer cells is associated with increased expression of nucleoside transporters and altered uptake of adenosine. Cancer Chemother Pharmacol. 1991;29:127-1232. [ Links ]
24. Chhikara BS, Mandal D, Parang K. Synthesis and evaluation of fatty acyl ester derivatives of cytarabine as anti-leukemia agents. Eur J Med Chem. 2010;45(10):4601-4608. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Jhon Fernando Berrío Escobar
Universidad de Antioquia (UdeA),
Calle 70 #52-21,
Medellín-Antioquia, Colombia.
jhon.berrio@udea.edu.co
+57-4-2198456
Received: 05-04-2016
Accepted: 12-05-2016

