










 Custom services
Custom services
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Triazolyl peptidyl penicillins (TAPs) are novel hybrids compounds with promising antitumoral activity.1,2 TAPs structures are constituted by a penicillanic core linked to a peptide portion via a triazole group. These previously reported structures were synthesized through a well stablished solid-phase methodology, that allowed to obtain a small library with good yields.
In a preceding study,2 we reported that derivative containing the dipeptide Leu-Phe (TAP7f, Figure 1), behaves as a selective and potent antitumor agent that induces an apoptotic response.3,4 In order to support downstream research activities with this candidate, we were initially requested to prepare increasing amounts of TAP7f. Herein we describe the strategy chosen to achieve that goal.

The solid-phase peptide synthesis has been widely used and developed since its description in 1963 by Merrifield.5 The methodology is based on attaching the amino acid, through the terminal carboxyl group, to an insoluble polymeric support and the following amino acids are subsequently coupled. The solid-phase peptides synthesis follows a general pattern of repetitive coupling-washing-deprotection-washing cycles.6
Some of the most important advantages of this methodology are: easy elimination of excess reagents, simplified purification by washing the solid support, and easy automation of the synthetic route. Solid-phase chemistry allows the generation of a wide variety of compounds, in a straightforward manner and in relatively short periods of time.7 Generally, this methodology is used to synthesize small amounts of many different compounds. When preclinical studies begin to be planned, larger quantities of the selected candidates are required. Unfortunately, for scale-up purposes, the common resins generally employed in solid-phase and combinatorial chemistry are quite expensive and impractical considering the waste of the polymer support. Therefore, from the economic point of view, the solution phase approach retains its usefulness for obtaining substances of interest, specially at a gram scale. Here, we present our solution phase strategy to obtain TAP7f in large scale.
RESULTS AND DISCUSSION
In the optimized previously reported solid phase synthesis of TAP7f1, Wang resin-bound Fmoc-phenylalanine (1, Scheme 1) was used as the starting material.
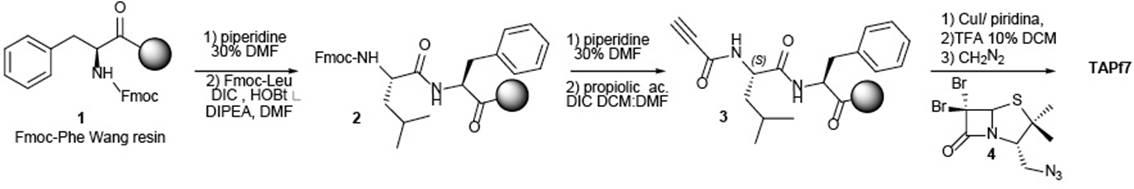
To replace this solid support, commercial enantiomerically pure L-phenylalanine was used. This is the step with the greatest impact in reducing the total cost of the process, since L-phenylalanine is approximately 45 times less expensive than the solid supported one.
TAP7f has its carboxylic acid group protected as methyl ester. This transformation was achieved with diazomethane at the final stage of the solid-phase strategy. Diazomethane reactions tends to be highly selectively, giving quite pure products in high yields, but on larger scales, the reaction is mostly avoided due to safety concerns.
Designing the synthesis process in solution phase, we realized that methylation of phenylalanine free acid had to be the first step to carried out to obtain TAP7f.
Thus, ester 6 (Scheme 2) was prepared treating L-Phe with 2 equiv of p-toluenesulfonic acid monohydrate in methanol,8 and the resulting solution was boiled under reflux for 18 h. Evaporation to dryness and trituration of the residue with ether yielded L-Phenylalanine methyl ester p-toluenesulfonate (6) as white solid. This salt was used for the next step without further purification.

Then, the coupling of protected leucine, the second amino acid, was carried out under standard conditions. For this step, Boc protecting strategy was chosen since it is cleavable under acidic conditions, releasing only volatile by-products. Synthesis of 7 was performed using only 1.1 equiv. of N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC:HCl) as the coupling agent, and 1-hydroxybenzotriazole (HOBt) as a racemization suppressant. In this way, less amount of reagents were used for the same reaction with respect to the solid phase strategy.
After Boc deprotection, optimization of the alkyne coupling to the molecule was the next challenge. Table 1 summarizes the results of the different reagents investigated. In the solid phase route, 20 equiv of DIC was used to the coupling step. To optimize and decrease the amounts of reagents used, we start testing dicyclohexylcarbodiimide (DCC)9 and diisopropylcarbodiimide (DIC)10 in lower amounts. Yields were good, but purification was complicated, due to the insoluble urea byproducts. When EDC.HCl11,12 was used as a coupling agent, yields were very low. For further optimization, we decided to explore the reaction with 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ).13 Fortunately, employing 1.1 equiv of this reagent, performance was similar to that obtained with DIC, but using smaller amounts of reagent and avoiding the undesirable urea byproducts.
Table 1. Different alkyne coupling conditions for the solution phase route
| Entry | Coupling reagent | Equiv. | solvent and other reagents | Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | DCC | 1.1 | DCM | 78* | 9 |
| 2 | DIC | 1.1 | DIPEA/DCM/DMF | 50* | 10 |
| 3 | EDC.HCl | 2 | DCM DIPEA | 7 | 11 |
| 4 | EDC.HCl | 2 | DCM/DMF/DIPEA | - | 12 |
| 5 | EEDQ | 1.1 | DCM | 66 | 13 |
* Urea by-products were difficult to remove
Then, the final stage involved the assembly of the penicillin azide 4 to the alkyne 8 with the Cu-catalyzed 1.3 dipolar cycloaddition. After 3 h of reaction, TAP7f was obtained with 74% for this final step, after chromatographic purification.
A comparison of the two synthetic strategies to obtain TAP7f is shown in Table 2.
Table 2. Comparison of the two synthetic strategies
| Entry | Topic | Solid phase synthesis | Solution phase synthesis |
|---|---|---|---|
| 1 | Steps | 7 | 5 |
| 2 | Global yield | 49 | 33 |
| 3 | Starting material cost (to synthesize 1g of product) | Fmoc-Phe Wang resin (4 g, 0.7mmol/g loading( U$D 530)* | L-Phe (0.8 g ( U$D 2)* |
| 4 | Aminoacid coupling reagents | HOBt (3 equiv.) EDC.HCl (3 equiv.) | HOBt (1.1 equiv.) EDC.HCl (1.1 equiv.) |
| 5 | Alkyne coupling step | DIC 20 (equiv) propiolic acid (10 equiv) | EEDQ (1.1 equiv) propiolic acid (1.1 equiv) |
| 6 | Time (h, global process) | 25.5 | 47.5 |
| 7 | Cromatographic purifications | 1 | 3 |
*obtained from Aldrich (catalog n°47666 and n°P2126)
With this new strategy, TAP7f can be prepared from a readily available, inexpensive starting material such as L-Phe and using less amounts of reagents for the intermediate steps, rendering this new process more amenable to the large-scale production.
EXPERIMENTAL
Solvents were analytical grade or were purified by standard procedures prior to use. Infrared spectra (IR) were recorded on a Shimadzu Prestige 21 spectrophotometer and only partial spectral data are listed. The 1H and 13C NMR spectra were acquired in the specified solvent, in a Bruker Avance spectrometer (300 and 75 MHz for 1H and 13C, respectively), with tetramethylsilane (TMS) as internal standard. The chemical shifts (δ) are reported in ppm downfield from TMS and coupling constants (J) are expressed in hertz. Flash column chromatography was performed using Merck silica gel 60 (230-400 mesh). Elution was carried out with hexane-EtOAc mixtures, under positive pressure and employing gradient of solvent polarity techniques.
Typical Procedure
L-phenylalanine methyl ester p-toluenesulfonate (6). A suspension of L-phenylalanine 5 (2.01 g, 12.16 mmol) in methanol (20 mil) was treated with p-toluenesulfonic acid monohydrate (4.52 g, 24.32 mmol), and the resulting solution boiled under reflux for 24 hours. Evaporation to dryness and trituration of the residue with ether followed by washing with ether yielded 6 (3.84 g, 90%), and it was used in the next step without further purification. RMN de 1H (CDCl3, 300MHz) δ 2.32 (s,CH3), 3.11(m, 2H,CH2), 3,46 (s, 3H, OCH3), 4,26 (m, 1H, CH), 7.00-7.71 (m, 9H arom.), 8.13 (s, 2H, NH3+), RMN de 13C (CDCl3, 75MHz) δ 21.3 (CH3), 36.9 (CH2), 52.9 (OCH3), 126.7 (CH), 127.5 (CH), 129.6 (CH), 134.6 (C), 140.8(C), 165.6 (CO), 169.4 (CO).
Methyl (tert-butoxycarbonyl)-L-leucyl-L-phenylalaninate (7). Compound 6 was dissolved in DCM and cooled to 0°C. Boc-Leu (1.1 equiv.), EDC.HCl (1.1 equiv), HOBt (1.1 equiv) and DIPEA (3.5 equiv) were added. The mixture was allowed to warm to room temperature and stirred for 24 h. After this time, a solution of NaHCO3 was added and the mixture was extracted with AcOEt (2x 30ml). The organic phase was dried with Na2SO4 and concentrated under high vacuum. The resulted crude product was purified by column chromatography (hexane:AcOEt) to yield 7(3.14g, 75%). RMN de 1H (CDCl3, 300MHz) δ 0.91 (m, 6H, 2 CH3), 1.43 (s, 9H, 3CH3), 1.52-1.66 (m, 2H, CH2), 3.12(m, 2H,CH2), 3,70 (s, 3H, OCH3), 4,06 (m, 1H, CH), 4.84 (m, 1H, CH), 7.08-7.31 (m, 5H arom.), 7.68 (s, 1H, NH), 8.03 (s, 1H, NH), RMN de 13C (CDCl3, 75MHz) δ 23.9 (CH3), 25.8 (CH3), 28.2 (CH3), 37.8 (CH2), 41.3 (CH2), 52.4 (OCH3), 53.1 (CH), 53.5 (CH), 80.7 (C), 136 (C), 155.6 (CO), 173 (CO).
Methyl propioloyl-L-leucyl-L-phenylalaninate (8). Compound 7 was deprotected with TFA:DCM (1:1) in 30 min. After evaporation, the residue was dissolved in DCM at 0°C. Then EEDQ (1,1 equiv), propiolic acid (1.1 equiv) and DIPEA (2 equiv) were added and the mixture was stirred for 2 h, and washed with 2.5 M HCl (2x30ml) and dried (Na2SO4). After filtration, the residue was purified by column chromatography (hexane:AcOEt) to give 8 (1.81g, 66%, 2 steps). RMN de 1H (CDCl3, 300MHz) δ 0.86 (m, 6H, 2CH3), 1.49 (m, 2H, CH2), 1.56 (m, 1H, CH), 2.05 (s, 1H, CH), 3.07(m, 2H,CH2), 3,70 (s, 3H, OCH3), 4,50 (m, 1H, CH), 4.80 (m, 1H, CH), 6.32 (s, 1H, NH), 6.69 (s, 1H, NH), 7.05-8.07 (m, 5H arom). RMN de 13C (CDCl3, 75MHz) δ 22.1 (CH3), 22.3 (CH3), 20.7 (CH), 25.4 (CH), , 38.7 (CH2), 41.1 (CH2), 51.6 (CH), 52.3 (OCH3), 53.2 (CH), 77.2 (C), 108 (CH), 120.6 (CH), 125.6 (CH), 128.7 (CH),135.5 (C), 143.5 (CO), 158.9 (CH), 163.5 (CO), 171.9 (CO).
Methyl(1-(((2S)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1azabicyclo [3.2.0]heptan-2-yl)methyl)-1H-1,2,3-triazole-4-carbonyl)-L-leucyl-L-phenylalaninate (TAP7f). Compound 8 was dissolved in THF, and CuI (0.5 equiv) DIPEA (5 equiv) and penicillin azide 4 (1.1 equiv) were added. The mixture was stirred for 3 h. After solvent evaporation, the residue was purified by column chromatography (hexane:AcOEt) to give TAP7f (2.79g, 74%). NMR spectral data of TAP7f were identical to those reported in the literature.2 1H NMR (CDCl3, 300 MHz) δ 0.91 (d, J= 5.6 Hz, 3H, CH3), 0.92 (d, J= 5.6 Hz, 3H, CH3), 1.57 (s, 3H, CH3), 1.60 (s, 3H, CH3), 1.59-1.76 (m, 3H, CH and CH2), 3.04 (dd, J1= 13.7 Hz, J2= 6.3 Hz, 1H, CH2), 3.14 (dd, J1= 13.7 Hz, J2= 5.7 Hz, 1H, CH2), 60 3.69 (s, 3H, OCH3), 4.37 (dd, J1= 13.3 Hz, J2= 10 Hz, 1H, CH2), 4.49 (dd, J1= 10 Hz, J2= 3.9, 1H, CH), 4.60 (m, 1H, CH), 4.70 (dd, J1= 13.3Hz, J2= 3.9, 1H, CH2), 4.87 (m, 1H, CH), 5.66 (s, 1H, CH), 6.75 (d, J= 8 Hz, 1H, NH), 7.04-7.15 (m, 5H, ArH), 7.38 (d, J= 8.2 Hz, 1H, NH), 8.26 (s, 1H, CH). 13C NMR (CDCl3, 75 MHz) δ 21.9 (CH3), 22.9 (CH3), 24.5 (CH), 24.6 (CH3), 33.3 (CH3), 37.9 (CH2), 40.7 (CH2), 48.4 (CH2), 51.3 (CH), 52.3 (CH), 53.2 (CH) 58.5 (C), 63.7 (C), 67.9 (CH), 78.4 (CH), 126.2 (CH), 127.0 (CH), 128.5 (CH), 129.2 (CH), 135.6 (C), 143.0 (C), 159.7 (CO), 165.6 (CO), 171.0 (CO), 171.8 (CO). IR: (film) 1797 cm-1 (β-lactam), 1744 cm-1 (CO ester), 1670 cm-1 (CO amide). HRMS-ESI (m/z): calcd for C27H34Br2N6NaO5S [M+Na]+: 735.05704; found: 735.05518.
CONCLUSION
In summary, an approach using solution phase chemistry for the synthesis of TAP7f has been developed. We have demonstrated that this new, five-step, procedure is a more efficient and economical route to synthesize the target molecule in large scale.
Full-scale optimization of the synthetic sequence is being investigated, especially to be able to eliminate chromatographic purifications by more convenient methods.

