










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkPuntos clave
Los artículos relacionados con las autorizaciones de medicamentos son abundantes, sin embargo, son escasos los referidos a los procedimientos usados por las Agencias Reguladoras del Medicamento en los diferentes Estados para emitir dichas autorizaciones y especialmente los utilizados durante la crisis sanitaria como la de la COVID-19.
Los estudios realizados sobre el tema en los Estados Unidos y la Unión Europea, están principalmente enfocados a la evaluación de la seguridad y eficacia de las vacunas frente a la COVID-19 y no tanto hacia el procedimiento utilizado para su aprobación. Los trabajos existentes sobre los procedimientos de autorización de estás vacunas, se limitan a una comparación del proceso antes y durante la emergencia sanitaria, sin desarrollar los aspectos relativos a los diferentes pasos en la tramitación del procedimiento de autorización como tal.
En el ámbito de América Latina, el estado de la cuestión es aún más limitada. Los trabajos que abordan el tema se enfocan principalmente al uso del reliance, sin hacer mayor referencia a los otros posibles mecanismos previstos por la OMS.
El presente, por tanto, pretende exponer la actuación de las Autoridades Regulatorias del Medicamento con relación a la autorización de las vacunas para la COVID-19, detallando para ello los procedimientos, la tramitación de éstos y los mecanismos empleados en el contexto de la pandemia.
Introducción
El reconocimiento por parte de la Organización Mundial de la Salud (OMS) de la enfermedad COVID-19 como pandemia el 11/03/20201, planteó un reto al mundo; no solo por el cambio en el estilo de vida de las personas, sino también en la forma de actuar de las Autoridades Reguladoras Nacionales del Medicamento (ARNs).
Han pasado más de 2 años de esta declaración, y aún la pandemia sigue vigente y en evolución constante. Solo en agosto del 2022, habíamos superado el millón de muertes por la COVID-19 causada por el SARS-CoV-2 y sus variantes.2
Desde diciembre de 2020 hasta la fecha, son varias las vacunas que se han autorizado y siguen autorizándose en el mundo por parte de las ARNs.
La autorización de las vacunas por procedimientos estándares, acorde a la legislación farmacéutica, debe ser obtenida, en principio, antes de su comercialización, de manera que asegure, ab initium, el cumplimiento de los criterios de calidad, seguridad y eficacia requeridos en los medicamentos. Para ello, las ARNs establecen procedimientos de evaluación que pueden llegar a superar los 12 meses, antes de que la ARN correspondiente emita una resolución sobre la autorización.
En una emergencia de salud pública como la COVID-19, el tiempo de respuesta es vital en la contención a la pandemia. Por consiguiente, las autorizaciones, en este contexto, deben ser lo suficientemente rápidas para ofrecer una respuesta a la pandemia sin afectar la calidad de la evaluación y aprobación de los instrumentos terapéuticos necesarios para luchar contra ella; es decir, en los nuevos productos debe mantenerse un equilibrio entre la valoración de la eficacia y la seguridad de éstos y la velocidad de la aprobación de los mismos.3
Respuesta a la pandemia de la COVID-19
El fundamento de la gobernanza global de una pandemia se encuentra en el Reglamento Sanitario Internacional (RSI), cuyo fin es “la prevención de la propagación internacional de enfermedades, proteger contra esa propagación, controlarla y darle una respuesta de salud pública proporcionada y restringida a los riesgos para la salud pública”.4
Es aquí donde los recursos terapéuticos, como es el caso de las vacunas, juegan un papel importante en la prevención de dicha propagación. Estos recursos son, sin duda, los más complejos de desarrollar, ya desde su planteamiento conceptual hasta la etapa de recopilación de evidencias suficientes de su calidad, seguridad y eficacia, que garanticen que su uso proporcionará más beneficios que riesgos.5
El RSI4, en el artículo 3, apartado 4, señala:
De conformidad con la Carta de las Naciones Unidas y los principios del derecho internacional, los Estados tienen el derecho soberano de legislar y aplicar leyes en cumplimiento de sus políticas de salud. Al hacerlo, respetaran la finalidad del presente reglamento.
De este modo emerge una referencia a la introducción de una legislación en la materia en cada uno de los Estados firmantes del RSI (entre ellos Estados Unidos, la Unión Europea y América Latina). La creación de mecanismos y procedimientos legales debe estar orientada a brindar una respuesta rápida a la emergencia. Su utilización, acorde con el RSI, esta supeditada a la declaración formal de una emergencia de salud pública de preocupación internacional por parte de la dirección de la OMS.4 Esta circunstancia concurrió con respecto a la COVID-19 el 30/01/2020, y fue la base para la posterior declaración como pandemia el 11/03/20201.
Dentro de esta normativa necesaria para afrontar una situación de pandemia, se encuentra la relativa al sector farmacéutico, pieza clave frente a una crisis sanitaria. De esta legislación, la referida a la autorización de comercialización de las vacunas para la COVID-19 toma un papel protagonista, debido a que su tarea es la optimización del tiempo del proceso de autorización y la conservación del balance beneficio-riesgo positivo en la eficacia y seguridad de los nuevos productos. La OMS6 señala que “en ese proceso es posible que no se cuente con el grado deseable de evidencia e información para tomar decisiones, razón por la cual la aplicación del principio precautorio resulta inevitable”. Siguiendo a Emilia Sánchez,7 “este principio puede describirse operativamente como la estrategia que, con enfoque preventivo, se aplica a la gestión del riesgo en aquellas situaciones donde hay incertidumbre científica sobre los efectos que en la salud (…) puede producir una actividad determinada.” Esta autora señala que, para su aplicación, se requiere que “se disponga de evidencia de que el riesgo que comporta es aceptablemente bajo y no solo de ausencia de evidencia de que el riesgo es elevado o inaceptable (…)”.
Esta expresión es una muestra de la complejidad de la toma de decisiones en la autorización o no de un nuevo producto, y más aún cuando se trata de un producto destinado a la prevención de una nueva patología que se propaga rápidamente y a niveles alarmantes, causando un alto porcentaje de muertes, y de la cual no se dispone de información suficiente o alternativas de tratamiento, como es el caso de la COVID-19.
Por este motivo, las ARNs, especialmente las que poseen mecanismos para autorizar el uso de productos en investigación, tuvieron que desarrollar pautas y procedimientos, crear grupos de trabajo y alianzas para maximizar la eficiencia de la evaluación, revisión y autorización de las vacunas.8 El objetivo del presente trabajo es la exposición de los procedimientos usados por las ARNs que permitieron el uso de las vacunas para la COVID-19, ello con el fin de determinar si la multiplicidad de procedimientos ha dado origen al registro de un elevado número de vacunas.
Métodos
Para la consecución del objetivo del trabajo, se realizó una revisión de la legislación farmacéutica aplicada a las autorizaciones de las vacunas para la COVID-19 emitidas durante la emergencia sanitaria. La información fue recopilada hasta el 15 de octubre de 2022 y extraída de sitios web gubernamentales de las ARNs, de:
Unión Europea: Agencia Europea del Medicamento (EMA - European Medicine Agency)
Estados Unidos de América (EE.UU.): Administración de Medicamentos y Alimentos (FDA - Food and Drug Administration)
-
América Latina:
Argentina: Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT)
Bolivia: Agencia Estatal de Medicamentos y Tecnologías en Salud (AGEMED)
Brasil: Agencia Nacional de Vigilancia Sanitaria (ANVISA)
Chile: Agencia Nacional de Medicamentos (ANAMED)
Colombia: Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA)
Costa Rica: Ministerio de Salud
Cuba: Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos (CECMED)
Ecuador: Agencia Nacional de Regulación, Control y Vigilancia Sanitaria (ARCSA)
El Salvador: Dirección Nacional de Medicamentos
Honduras: Agencia de Regulación Sanitaria (ARSA)
Guatemala: Departamento de Regulación y Control de Productos Farmacéuticos y Afines
México: Comisión Federal para la Protección Contra Riegos Sanitarios (COFEPRIS)
Nicaragua: Dirección General de Regulación Sanitaria
Panamá: Dirección General de Farmacias y Drogas
Paraguay: Dirección Nacional de Vigilancia Sanitaria (DINAVISA)
Perú: Dirección General de Medicamentos, Insumos y Drogas (DIGEMID)
Uruguay: Departamento de Medicamentos
Venezuela: Dirección de Drogas, Medicamentos y Cosméticos.
También se incluyo información obtenida de otras fuentes, tales como artículos de revistas científicas e información de sitios web oficiales como la OMS y la Organización Panamericana de la Salud (OPS).
Resultados y Discusión
Estados Unidos: FDA
La Autorización del Uso de Emergencia (EUA - Emergency Use Authorization), es el mecanismo por el cual la FDA permite la comercialización de productos no aprobados o productos aprobados para usos no aprobados y cuyo empleo esta supeditado a situaciones de emergencia de salud pública previamente declaradas por la autoridad correspondiente del país; por tanto, su uso no representa una aprobación del producto en el sentido legal del término, sino su uso y comercialización en las circunstancias antes descritas.
La Federal Food, Drug and Cosmetic Act, sección 564 Emergency Use Authorization9, es la base normativa de la EUA. Esta señala, que su uso se justifica, entre otras, con:
Una determinación por el Secretario [de Salud y Servicios Humanos] de que hay una emergencia de salud pública o una potencial emergencia de salud pública significativa, que afecta o tiene un potencial significativo para afectar la seguridad nacional o la salud (…), y que involucra un agente o agentes biológicos, químicos, radiológicos o nucleares, o una enfermedad o condición que pueda ser atribuible a tal agente o agentes.
Evento que ocurrió el 31/01/2020, cuando se declaró que la COVID-19 representaba una amenaza a la salud pública; posteriormente el 04/02 en conformidad con la sección 564 se declaró la emergencia sanitaria que involucró al virus que causa la COVID-19. Con base en esta determinación el 27/03/2020 el Secretario declaró que existían circunstancias que justificaban la autorización de uso de emergencia de medicamentos y productos biológicos durante la pandemia de la COVID-19.10
La EUA es considerada una herramienta de respuesta rápida ante la emergencia de salud pública, por tanto, su validez está relacionada con la duración de dicha emergencia.
Las vacunas para la COVID-19, usadas en los EE.UU., mantienen su vigencia a fecha de hoy, debido a que el país no ha dado por finalizada la emergencia de salud pública; al contrario, ésta ha sido renovada hasta en 11 ocasiones y su última renovación, de fecha 13/10/2022 mantiene la emergencia de salud pública vigente.11
El mecanismo usado para autorizar las vacunas para la COVID-19, acorde a la legislación es el correspondiente a los productos no aprobados por tratarse de moléculas nuevas. El proceso de emisión por esta vía se detalla en la Fig. 1.

El proceso de obtención se inicia con una solicitud formal; sin embargo, la FDA considerando la urgencia de tratamientos médicos ante una emergencia sanitaria, establece una etapa denominada actividades Pre-EUA, que está definida como la interacción preliminar entre la FDA y la industria. Se trata de una actividad de un solo paso, cuyo fin es discutir la idoneidad del producto potencial.12 No es obligatoria, pero si recomendable. La FDA señala que esta interacción debe realizarse lo antes posible.10 El medio sugerido para la solicitud Pre-EUA, es su consideración como Producto Nuevo en investigación (Pre-IND - pre- Investigational New Drug )13, ya que ello facilita la revisión y evaluación de la documentación presentada. El resultado de este paso no implica necesariamente la opinión de la FDA sobre el producto potencial o que el solicitante haya obtenido o presentado toda la información necesaria para que la FDA revise una solicitud formal de EUA.12
Terminada esta fase previa de actividades Pre-EUA (en caso de que el proceso se haya iniciado con dicho paso), se continua con la solicitud formal dirigida al centro correspondiente de la FDA. La solicitud ha de presentarse mediante el eCTD (Electronic Common Technical Document) junto con toda la información técnica disponible.
El siguiente paso en el proceso es la revisión por parte de la FDA de la información proporcionada por el solicitante. La decisión de revisar y procesar una solicitud se basa en una determinación caso por caso de que dicha acción es necesaria para proteger la salud pública en una emergencia. En esta etapa aparece el término puede ser efectivo, utilizado por la FDA para este tipo de autorizaciones, debido a que el nivel de evidencia es más bajo que el modelo de efectividad que usa para las autorizaciones de comercialización. La revisión y evaluación incluye sesiones abiertas o cerradas (usado en temas de propiedad intelectual) del Comité Asesor de Vacunas y Productos Biológicos, siendo éstas específicas para cada vacuna; el fin es discutir los datos se seguridad y eficacia disponibles que respaldan la EUA.10 La evaluación concluye con un dictamen del comité (realizada por votación entre los miembros); si éste es positivo, da lugar a la emisión de la EUA. Es importante señalar que, si se dejara de cumplir alguno de los criterios de la aprobación, esta autorización puede ser revocada, incluso antes de que finalice la declaración de emergencia.9
El plazo para llevar a cabo la evaluación no esta señalado en la legislación, sin embargo, en la guía Emergency use of medical products and related authorities, se menciona que la FDA está preparada para resolver de manera expedita dentro de horas o días.12 La vacuna de Pfizer empleó 21 días14, Moderna 18 días15, Janssen 85 días16 y Novavax 163 días.17
Con la emisión de la EUA, la FDA puede establecer condiciones que deban cumplirse por el titular de la autorización con el fin de proteger la salud pública (Ver Fig.1). Además de estas condiciones, los productos autorizados están sometidos a: 1) un plan de vigilancia activa (monitorización post-autorización) bajo responsabilidad del gobierno federal (ver Fig.1) y 2) una revisión periódica de las circunstancias y la pertinencia de la EUA. El objetivo de ambos condicionamientos es confirmar y mantener la relación beneficio- riesgo positiva del producto aprobado mediante este tipo de autorización.
Las enmiendas a la autorización de la vacuna Pfizer-BioNTech BNT162b2, son una muestra de esta revisión: la EUA inicial del 11/12/2020, para uso en personas mayores de 16 años, fue re-emitida 18 veces, con el fin de incluir modificaciones, como por ejemplo la inclusión de nuevos grupos de edad (12-15 años, 5-11 años, 6 meses a 4 años), entre otras.18 La autorización de refuerzos bivalentes del 31/08/2022 de las vacunas de Moderna y Pfizer-BioNTech frente a las variantes del SARS-Cov-2, como refuerzos actualizados y que eliminó el uso de las vacunas monovalentes de ambos fabricantes como dosis de refuerzo, manteniendo su uso solo para la administración primaria19, es una muestra más de dicha revisión.
Finalmente, la normativa americana señala la posibilidad que la EUA pueda convertirse en una autorización de comercialización en firme, ello supeditado al cumplimiento y presentación de los estudios requeridos por la FDA al efecto. La ARN de los EE.UU. no considera la obtención de una EUA como motivo para detener el seguimiento de un ensayo clínico en curso.10
Ejemplo de este cambio de autorización, lo supone la vacuna de Pfizer-BioNTech, BNT162b2, que con fecha de 23/08/2021 obtuvo por parte de la FDA, su licencia (autorización) de comercialización, bajo el nombre de Comirnaty para su uso en personas mayores de 12 años. Es importante mencionar que, de forma paralela a esta licencia, la FDA mantiene una EUA para esta vacuna, para usos que no están incluidos en la misma, tales como: serie de 2 dosis para personas de 5 años y mayores, un esquema de tercera dosis del esquema principal de vacunación para personas de 5 años y mayores que tienen cierto tipo de inmunodepresión, y como una serie primaria de tres dosis para personas de 6 meses a 4 años de edad.20
Unión Europea: EMA
El mecanismo empleado en el ámbito comunitario es la Autorización de Comercialización Condicional (ACC). La ACC, a diferencia de la EUA, es una autorización de comercialización condicionada a que se otorgue información adicional después de la comercialización del medicamento por parte de la empresa solicitante.
En línea con el RSI, la Decisión 1082/2013/UE sobre amenazas transfronterizas graves para la salud y el Plan de la EMA para amenazas sanitarias emergentes, son los documentos de activación de la ACC. Su aplicación está determinada por la existencia de dos supuestos: a) Determinación de una Emergencia de Salud Pública de Importancia Internacional por la OMS o la Comisión Europea; y b) Declaración de pandemia por la OMS o la Comisión Europea durante el período de propagación de la gripe humana causada por un nuevo subtipo. Ambas situaciones permiten la aplicación de diferentes niveles de activación, que se traducen en niveles diferentes de intervención del personal de la EMA en la emergencia.21,22
La pandemia de la COVID-19, acorde con el Plan, condujo automáticamente a una crisis de nivel 4, desencadenando la activación de todos los grupos de trabajo disponibles, personal de apoyo adicional y reasignación de recursos, con el fin de cubrir la necesidad potencial de autorizar medicamentos rápidamente22. Todo ello permitió aprovechar la experiencia de la red europea de regulación de medicamentos para garantizar una respuesta rápida a la pandemia.23
La base legal de la ACC es el Reglamento (CE) Nº 507/2006 de la Comisión de 29/03/2006 sobre la Autorización Condicional de Comercialización de los Medicamentos de Uso Humano que entran en el ámbito de aplicación del Reglamento (CE) Nº 726/2004 del Parlamento Europeo y del Consejo. Éste señala en su artículo 224 las condiciones para obtener una ACC:
-
Estar dentro del ámbito del artículo 3, apartados 1 y 2 del Reglamento (CE) Nº 726/2004 del Parlamento Europeo y del Consejo de 31/03/2004 por el que se establecen procedimientos comunitarios para la autorización y el control de medicamentos de uso humano y veterinario (…).
Este requerimiento implica el uso del procedimiento centralizado, para lo cual, el producto potencial debe encontrarse dentro de alguna de las categorías establecidas en el anexo de dicho reglamento (medicamentos obtenidos por procesos biotecnológicos, medicamentos con sustancia activa nueva para determinadas enfermedades -entre ellas las víricas-, medicamentos huérfanos).
Pertenecer a cualquiera de las siguientes categorías: a) medicamentos destinados al tratamiento, la prevención o el diagnóstico de enfermedades gravemente debilitantes o potencialmente mortales; b) medicamentos destinados a situaciones de emergencia en respuesta a amenazas para la salud pública debidamente reconocidas por la OMS o por la Comunidad Europea (…); c) medicamentos declarados huérfanos (…).
Todas las vacunas para la COVID-19 autorizadas por la EMA, utilizaron el procedimiento centralizado. El proceso de emisión de la ACC se detalla en la Fig. 2.

El proceso de obtención de la ACC se inicia con una solicitud formal. No obstante, al igual que la FDA, la EMA también realiza actividades previas (no obligatorias, pero recomendadas) a dicha solicitud, tales como: 1) el asesoramiento científico rápido y 2) la revisión continuada (rolling review). El objetivo de estas actividades es apoyar la planificación prospectiva de la generación de la evidencia científica del producto potencial y la revisión de los datos a medida que estos estén disponibles25, respectivamente. Muestra de la no obligatoriedad de estas actividades previas es la inexistencia del asesoramiento científico rápido en el caso de la vacuna Pfizer.26
El rolling review, a diferencia del asesoramiento científico, fue empleado en todas las vacunas para la COVID-19 como actividad previa. Está actividad se ejecuta por ciclos de revisión (el número de ciclos y el tiempo empleado en ellos depende de la información que se vaya a evaluar). Por ejemplo, la vacuna Moderna tuvo 1 ciclo de revisión27,28 y Nuvaxovid 429. El tiempo estimado empleado, según el trabajo realizado por Marinus Roelie et al, varía entre 16 a 46 días, con una duración media de 20 días.27
En contraste con la actividad pre-IND de un solo paso y sin conclusiones vinculantes de la FDA para la solicitud de una EUA, el asesoramiento científico rápido y el rolling review empleados por la EMA, contienen flujos definidos de varios pasos, como puede observarse en las Fig. 3 y 4. El rolling review por su parte, concluye con una opinión favorable (o no) y justificativa para la presentación de la solicitud formal de ACC.
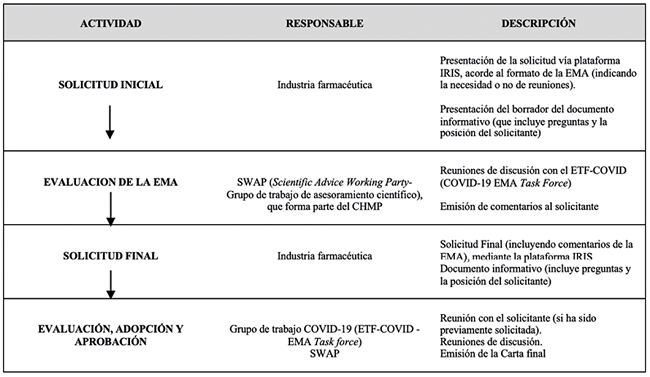
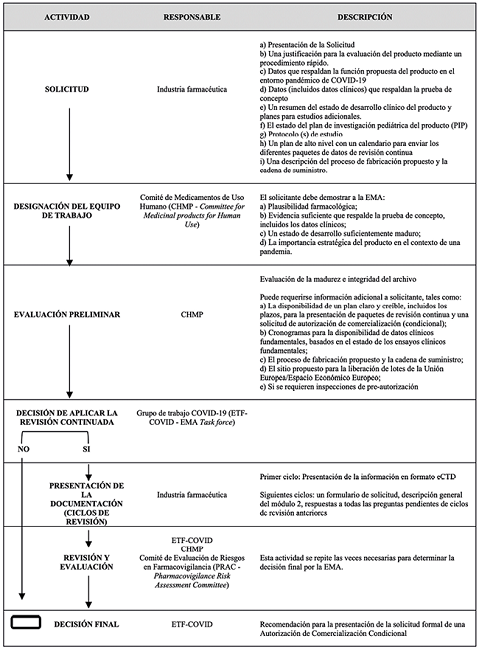
Finalizada la fase de actividades previas (en el supuesto de que se haya iniciado por ese paso), se pasa a la fase de formalización de la solicitud, a través del eCTD, esta solicitud va acompañada de los requisitos establecidos en la legislación correspondiente y de una justificación que señale: a) la necesidad médica no cubierta, b) la cuantificación de la necesidad médica insatisfecha teniendo en cuenta la argumentación técnica, y c) la medida en que el medicamento abordará la necesidad médica no satisfecha y sus ventajas para la salud pública.30
La siguiente fase del proceso es la evaluación por parte de la EMA acorde al Reglamento (CE) Nº 726/2004 (ver Fig.2). En esta etapa el producto debe: 1) demostrar una relación beneficio-riesgo positiva, 2) La posibilidad de suministrar la totalidad de los datos clínicos, 3) Satisfacer las necesidades médicas no cubiertas y 4) Demostrar que las ventajas para la salud pública que se derivan de la disponibilidad inmediata del medicamento en cuestión en el mercado son superiores al riesgo inherente a que todavía se necesiten más datos.24,30
La evaluación de la ACC no tiene marcado un plazo definido como la autorización de comercialización, pero se asume que debe ser menor en atención al contexto en el que se aplica. El tiempo empleado por la EMA para este paso, no ha superado los 40 días naturales: AstraZeneca 17 días31, Moderna 36 días28, Jansen 25 días32, Pfizer 20 días26; la excepción es Nuvaxovid con 48 días29.
Finalizada la evaluación y emitida la opinión del Comité de Medicamentos de Uso Humano (CHMP), si ésta es positiva, se procede a la emisión de la ACC (conjuntamente con las obligaciones específicas que deben cumplirse por parte del solicitante), con validez de 1 año.24
Asumiendo la validez de la ACC, el proceso se reanuda con su renovación (ver Fig.2). Si durante esta etapa, los datos incluidos justifican una actualización de la información del producto o del plan de gestión de riesgos, dichos cambios pueden formar parte del procedimiento de renovación. Se debe tener en cuenta que la renovación no reemplaza otras presentaciones requeridas (por ejemplo, variaciones) y que la presentación de dichos datos no debe posponerse para la próxima renovación.30 La EMA respecto de estas presentaciones requeridas, aplica el Reglamento (CE) Nº1234/2008 del 24/11/2008 relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización de medicamentos para uso humano y medicamentos veterinarios.33 La inclusión de la dosis de refuerzo para la variante omicron BA.1 del 01/09/2022 para las vacunas de Pfizer y Moderna34,35, es un ejemplo de ello.
El cumplimiento de las obligaciones específicas de una ACC, acorde al Reglamento Nº 507/2006, otorga la posibilidad de convertir dicha ACC en una autorización de comercialización convencional (al igual que la EUA de la FDA), siempre que esto sea recomendado por el CHMP. Este cambio puede realizarse en el momento de la renovación o en la evaluación de los datos presentados para cumplir la última obligación específica restante.30 Esto ha sucedido con las vacunas de Pfizer y Moderna, que han obtenido recientemente su autorización de comercialización al finalizar el cumplimiento de sus obligaciones especificadas.36
Finalmente, es importante señalar que de las 6 vacunas para la COVID-19 autorizadas en la Unión Europea (Pfizer, Moderna, Nuvaxovid, AstraZeneca, Janssen, Valneva), solo una de ellas (Valneva) obtuvo directamente la autorización de comercialización por parte de la EMA el 23/06/202237,38, sin necesidad de obtener previamente una ACC.
América Latina: ARNs
Al igual que EE.UU. y la Unión Europea, en América Latina la declaración de la COVID-19 como pandemia, generó una serie de declaraciones de estado de emergencia. Dichas declaraciones fueron emitidas por el Presidente de la República de cada país mediante los procedentes instrumentos legales, tales como: leyes, decretos, ordenanzas, resoluciones o acuerdos ministeriales, como se muestra en la Tabla 1.
Tabla 1. Declaración de emergencia en los países de América Latina.
| PAÍS | BASE LEGAL DE LA DECLARACIÓN DE EMERGENCIA |
|---|---|
| ARGENTINA | Decreto de Necesidad y Urgencia Nº 260/2020 del 12/03/2020, dispuso ampliar la Emergencia pública en materia sanitaria establecida por Ley Nº 27541, en virtud de la pandemia declarada por la OMS en relación al COVID-19 |
| BOLIVIA | Decreto Supremo Nº 4196 del 17/03/2020 que declara la emergencia sanitaria Nacional |
| BRASIL | Ordenanza No. 188 de 3 de febrero de 2020, que declaró Emergencia en Salud Pública de Importancia Nacional (ESPIN), como consecuencia de casos sospechosos de Infección Humana por el nuevo Coronavirus (SARS-CoV-2) |
| CHILE | Decreto 4, de fecha 08 de febrero de 2020, que, Decreta Alerta Sanitaria por el período que se señala y otorga facultades extraordinarias que indica por emergencia de salud pública de importancia internacional (ESPII) por brote del nuevo coronavirus (2019-NCOV) |
| COLOMBIA | Resolución 385 de 12 de marzo de 2020, Por la cual se declara la emergencia sanitaria por causa del coronavirus y se adoptan medidas para hacer frente al virus |
| COSTA RICA | Decreto Ejecutivo 42227-MP-S, del 16 de marzo de 2020, que declara el estado de emergencia nacional en todo el territorio de la República de Costa Rica, debido a la situación de emergencia sanitaria provocada por la enfermedad causada por el COVID-19. |
| CUBA | Resolución 82 de 23 de marzo de 2020, emitida por el Ministerio de Salud Pública manifiesta la situación epidemiológica de emergencia ante la presencia en el país de la COVID-19 |
| ECUADOR | Acuerdo Ministerial Nº 00057-2020, de 14 de septiembre de 2020 Disponer de la emergencia del Sistema Nacional de Salud a fin que se mantengan las medidas necesarias para garantizar el derecho a la Salud en la población ante la crisis sanitaria existente provocada por el SARS CoV-2 |
| EL SALVADOR | Decreto Legislativo Nº 593, de 14 de marzo de 2020 Estado de Emergencia Nacional de la Pandemia por COVID-19 |
| HONDURAS | PCM-005-2020, del 10 de febrero de 2020, por el que se Declara el estado de emergencia sanitaria por COVID-19 |
| GUATEMALA | Decreto Gubernativo Nº 5-2020, del 5 de marzo de 2020 Declarar en estado de calamidad pública en todo el territorio como consecuencia del pronunciamiento de la OMS de la epidemia del COVID-19 como emergencia de salud pública de importancia internacional y del Plan par la Prevención, Contención y Respuesta a casos de COVID-19 en Guatemala del Ministerio de Salud Pública y Asistencia Social. |
| MÉXICO | 30 de marzo de 2020. Acuerdo por el que se declara como emergencia sanitaria por causa de fuerza mayor, a la epidemia de enfermedad generada por el virus SARS-CoV2 (COVID-19) |
| NICARAGUA | |
| PANAMÁ | Resolución de Gabinete Nº 11 de 13 de marzo de 2020, Que declara el Estado de Emergencia Nacional y dicta otras disposiciones |
| PARAGUAY | Ley Nº 6524 del 25 de marzo de 2020 Que declara Estado de emergencia en todo el territorio de la República de Paraguay ante la pandemia declarada por la OMS a causa del COVID-19 o coronavirus y establece medidas administrativas, fiscales y financieras. |
| PERÚ | Decreto Supremo Nº 044-2020-PCM, de 15 de marzo de 2020 Que declara Estado de Emergencia Nacional por las graves circunstancias que afectan la vida de la Nación a consecuencia de brote del COVID-19. |
| URUGUAY | Decreto Nº 93/020 de 13 de marzo de 2020 Declaración de Estado de Emergencia Nacional Sanitaria como consecuencia de la pandemia originada por el virus COVID-19 (coronavirus) |
| VENEZUELA | Decreto Nº 4160 de 13 de marzo de 2020 Decreta el estado de Alarma en todo el territorio nacional, para atender la Emergencia Sanitaria del Coronavirus (COVID-19). |
Es importante señalar que sólo en los casos de Brasil, Chile, Honduras y Guatemala, fueron emitidas con anterioridad al 11/03/2020; estos países justificaron esta decisión en la declaración de la OMS que consideraba a la COVID-19 como una emergencia de salud pública de preocupación internacional. Nicaragua fue el único país en América Latina que no aplicó decretos ejecutivos de emergencia nacional ni ningún otro marco regulatorio39, tal como se puede ver en las Tablas 1 y 2.
Tabla 2. Mecanismos de aprobación de las vacunas para la COVID-19 y su base legal en América Latina.
| PAÍS | AUTORIDAD REGULADORA DEL MEDICAMENTO | M1 | M2 | M3 | M4 | BASE LEGAL |
|---|---|---|---|---|---|---|
| ARGENTINA | Administración Nacional de Medicamentos, Alimentos y Tecnología Médica - ANMAT | ✓ | ✓ | - | - | Ley 27573 del 06 de noviembre de 2020 Ley de vacunas destinadas a generar inmunidad adquirida contra el COVID-19 y sus modificatorias. |
| BOLIVIA | Agencia Estatal de Medicamentos y Tecnologías en Salud - AGEMED | - | ✓ | - | - | D.S. Nº 4432 del 29/12/2020, que tiene por objeto autorizar a las entidades competentes la contratación directa, bajo los principios de transparencia y legalidad, de vacunas, pruebas diagnósticas, medicamentos, dispositivos médicos, insumos, reactivos, equipamiento médico, así como otros bienes, obras y servicios, para la contención, diagnóstico y atención de la COVID-19. D.S. Nº 4438 del 30/12/2020, cuyo objetivo es establecer los requisitos que deben cumplir los proveedores de vacunas contra la COVID-19 en el mercado interno. Ley 1359 del 17/02/2021 Ley de Emergencia Sanitaria. |
| BRASIL | Agencia Nacional de Vigilancia Sanitaria - ANVISA | - | ✓ | ✓ | - | Ley 14124 del 10 marzo de 2021 Dispone medidas excepcionales en materia de adquisición de vacunas e insumos y contratación de bienes y servicios de logística, tecnologías de la información y comunicación, comunicación social y publicidad y capacitación para la vacunación contra el covid-19 y sobre el Plan Nacional de Operacionalización de la Vacunación contra el Covid-19. Resolución 475, del 10 de marzo de 2021, que establece los procedimientos y requisitos para solicitar un pedido de autorización temporal de uso de emergencia con carácter experimental de medicamentos y vacunas para covid-19. Instrucción Normativa Nº 77, del 17 de noviembre de 2020 Disposición sobre el procedimiento de solicitud continua de datos técnicos para el registro de vacunas covid-19. Resolución Directiva Colegiada Nº 415 de 26 de agosto de 2020: Define los nuevos criterios y procedimientos extraordinarios para tratamiento de peticiones de registro y traslado post registro de medicamentos y productos biológicos en virtud de la emergencia de salud publica internacional del nuevo coronavirus. |
| CHILE | Agencia Nacional de Medicamentos - ANAMED | ✓ | ✓ | - | - | Decreto 65 de 21 de agosto de 2020. Modifica el Decreto Supremo Nº 3, de 2010, del Ministerio de Salud, que aprueba el Reglamento del Sistema Nacional de Control de los Productos Farmacéuticos de uso humano en materia de equivalencia terapéutica. |
| COLOMBIA | Instituto Nacional de Vigilancia de Medicamentos y Alimentos - INVIMA | ✓ | ✓ | - | - | Decreto Nº 1787 de 29 de diciembre de 2020, Por el que se establecen las condiciones sanitarias para el tramite y otorgamiento de la autorización sanitaria de uso de emergencia - ASUE para medicamentos de síntesis química y biológicos destinados al diagnóstico, la prevención y tratamiento de la Covid-19 en vigencia de la emergencia sanitaria. |
| COSTA RICA | Ministerio de Salud | - | ✓ | - | - | DM-RM-7905-2020 del 03 de diciembre de 2020. Resolución administrativa de autorización de uso de las vacunas contra COVID-19 basado en el reconocimiento de la autorización de comercialización o autorización de uso de emergencia de autoridades reguladoras estrictas o precalificadas por la OMS. DM-RM-0933-2021 del 26 de abril de 20201 Resolución administrativa de autorización de uso e importación de las vacunas contra COVID-19 en el sector privado basado en el reconocimiento de la autorización de comercialización o autorización de uso de emergencia de autoridades reguladoras estrictas o precalificadas por la OMS o en la lista de uso de emergencia de la OMS. |
| CUBA | Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos - CECMED | ✓ | - | - | - | Resolución Nº 54 de 29 de mayo de 2020, Autorización de uso de emergencia de medicamentos y productos biológicos de uso humano, dispositivos médicos y otras tecnologías sanitarias, ante eventos de situaciones de emergencia declaradas por las autoridades competentes. Resolución Nº 57, de fecha 13 de julio de 2020, que puso en vigor la Regulación Nº M-75-20 Requisitos para la solicitud de Autorización de uso de emergencia de medicamentos y productos biológicos de uso humano en investigación. |
Tabla 2 (continuación). Mecanismos de aprobación de las vacunas para la COVID-19 y su base legal en América Latina (continuación).
| PAÍS | AUTORIDAD REGULADORA DEL MEDICAMENTO | M1 | M2 | M3 | M4 | BASE LEGAL |
|---|---|---|---|---|---|---|
| ECUADOR | Agencia Nacional de Regulación, Control y Vigilancia Sanitaria - ARCSA | - | ✓ | - | - | Ley 67 Ley Orgánica de Salud, modificada por la séptima decisión transitoria de la Ley Orgánica de incentivos para asociaciones público privadas y la inversión extranjera. Resolución Nº ARCSA-DE-016-2020-LDCL Expídase la normativa técnica sustitutiva para autorizar la importación por excepción e importación por donación de medicamentos, productos biológicos, dispositivos médicos y reactivos bioquímicos y de diagnóstico. Resolución Nº ARCSA-DE-037-2020-LDCL Expedir la reforma parcial a la normativa técnica sustitutiva para autorizar la importación por excepción e importación por donación de medicamentos, productos biológicos, dispositivos médicos y reactivos bioquímicos y de diagnóstico. |
| EL SALVADOR | Dirección Nacional de Medicamentos | - | ✓ | - | - | Resolución Nº 333-2013, del 12 de diciembre de 2013 Aprueba RTCA 11.03.59.11 Medicamentos Uso Humano. Registro Sanitario. |
| HONDURAS | Agencia de Regulación Sanitaria - ARSA | - | ✓ | - | - | C-001-ARSA 2021 Reconocimiento de vacunas SARS-CoV-2. |
| GUATEMALA | Departamento de Regulación y Control de Productos Farmacéuticos y Afines | - | ✓ | - | - | Decreto Nº 1-2021, de 23 de enero de 2021 Ley para el financiamiento y adquisición de vacunas contra el COVID-19. |
| MÉXICO | Comisión Federal para la Protección Contra Riegos Sanitarios - COFEPRIS | ✓ | ✓ | - | - | DOF 11/11/2020: ACUERDO por el que se instruyen a la Secretaría de Salud y a la Comisión Federal para la Protección contra Riesgos Sanitarios las acciones que en el mismo se indican. |
| NICARAGUA | Dirección General de Regulación Sanitaria | - | - | - | - | - |
| PANAMÁ | Dirección General de Farmacias y Drogas | - | ✓ | - | - | Resolución Nº 037, de 17 de febrero de 2021 Que establece el procedimiento para la emisión de la Autorización de Uso de Emergencia (AUE) de las vacunas contra el SARS-CoV-2 y los requisitos para la autorización de importación de las mismas. |
| PARAGUAY | Dirección Nacional de Vigilancia Sanitaria - DINAVISA | - | ✓ | - | - | Resolución S. G. N° 746, de 29 de diciembre de 2020 Por el cual se autoriza en carácter de emergencia vacunas contra el COVID-19. Resolución S. G. Nº 0111, de 04 de marzo de 2021. Por la cual se establecen los requisitos y condiciones sanitarias para la emisión de registros sanitarios de emergencia para especialidades farmacéuticas contra COVID-19, en el marco de pandemia del coronavirus. |
| PERÚ | Dirección General de Medicamentos, Insumos y Drogas - DIGEMID | - | ✓ | - | - | Ley Nº 31091 De 18 de diciembre de 2020 Ley que garantiza el acceso al tratamiento preventivo y curativo de la enfermedad por coronavirus SARS-COV-2y de otras enfermedades que dan origen a emergencias sanitarias nacionales y otras pandemias declaradas por la OMS. D.S. Nº 002 -2021 S.A. Del 08 de enero de 2021 Que aprueba el Reglamento para el Registro Sanitario Condicional de Medicamentos y productos Biológicos y modificatorias. |
| URUGUAY | Departamento de Medicamentos | - | ✓ | - | - | Decreto Nº 18/020 de13 de enero de 2020 Reglamento para el registro, producción, exportación, importación y comercialización de medicamentos de uso humano. |
| VENEZUELA | Dirección de Drogas, Medicamentos y cosméticos | - | - | - |
Con el fin de asegurar la disponibilidad de productos terapéuticos, las ARNs de América Latina priorizaron los procesos de evaluación y aprobación, utilizando para ello mecanismos flexibles,40) presentes en su legislación.
La terminología utilizada en cada Estado para referirse a las autorizaciones usadas en el contexto de emergencia es diversa: autorización condicional, de emergencia, temporal, licencia especial de emergencia, certificado de uso de emergencia, registro sanitario condicional, entre otros. Pero en línea con la OPS, utilizaremos el término de “autorización de uso de emergencia”, para referirnos a todas ellas.41
Los mecanismos utilizados por las ARNs para la introducción de vacunas frente a la COVID-19, acorde con la OPS son: el mecanismo excepcional con base a reconocimiento (reliance), la autorización de uso de emergencia, el registro con condiciones normales y el procedimiento abreviado.42
a. Reconocimiento (reliance)
La OMS43 lo define como “el acto mediante el cual una ARN de una jurisdicción puede tener en cuenta y dar un gran peso a las evaluaciones realizadas por otra ARN o institución confiable para tomar su propia decisión”. Esté último hace alusión a la OMS y su Listado de Uso de Emergencia.44
Es potestad de cada ARN y cada gobierno tomar la determinación de lo que constituye una autoridad o institución confiable de referencia. 41
Por ejemplo, ARCSA (Ecuador), en la disposición Única de la Resolución Nº ARCSA-DE-037-2020LDCL45, establece que:
(…) ARCSA aceptará como registro sanitario o su equivalente la autorización de uso por emergencia o documento equivalente, emitido en el país de origen del producto o proveniente de un país cuya Agencia Reguladora Nacional (ARN´s) sea reconocida como de Alta Vigilancia Sanitaria por la OMS/OPS.
Esta premisa señala, por un lado, a la lista de autoridades reguladoras estrictas de la OMS46 y por otro, a las ARNs de referencia nivel IV de la OPS (ARN competente y eficiente en el desempeño de las funciones de regulación sanitaria)47. En esta última categoría, en América Latina se cuentan las ARN de Argentina, Brasil, Chile, Colombia, Cuba y México.
La mayoría de las ARNs de América Latina basan sus listados de autoridades de referencia en las recomendaciones de la OMS/OPS. Sin embargo, al ser potestad de cada Estado establecer su listado de ARNs de referencia, se pueden incluir otras ARN no recomendadas por la OMS. Un ejemplo de ello lo constituye, Brasil que incluye entre otras ARNs de referencia, al Ministerio de Salud de la Federación Rusa, la Administración Nacional de Productos Médicos de la República popular China, la Organización Central de Control de Normas y Drogas de la República de India, y a la Agencia de Prevención y Control de Enfermedades de Corea (artículo 16 de la Ley 14124)48.
Tal como se ha señalado ut supra, las ARNs pueden considerar también instituciones de referencia, como la OMS. Muestra de ello es la Resolución 037 del Ministerio de Salud de Panamá, en la que se señala, entre otros, el uso del listado de uso de emergencia de la OMS.49
La mayoría de ARNs de América Latina, especialmente las que no son consideradas de referencia regional, utilizaron este mecanismo, tal como se muestra en la Tabla 2. Esta observación es similar a lo señalado en el estudio de Van der Zee et al.50
Además, en este punto debemos señalar que países como Brasil, Chile y Honduras, que antes de la pandemia no aceptaban ninguna forma de dependencia para la emisión de una autorización de comercialización51, emitieron normativas relacionadas a este mecanismo de autorización en el contexto de pandemia de la COVID-19 (ver Tabla 2).
En cuanto a la aplicación del reliance por parte de las ARNs de América Latina para la emisión de una autorización de emergencia, debemos indicar que ésta puede ser total o parcial; esto quiere decir que se puede aceptar de manera automática la decisión de la ARN o institución de referencia, o se puede aceptar como requisito para acelerar el tiempo de revisión para la emisión de la autorización de emergencia. Ejemplo del uso de un reliance total lo encontramos en el caso de Bolivia, que en el Decreto Supremo Nº 443252, establece:
(…) los registros sanitarios emitidos por al menos una autoridad regulatoria de alta vigilancia de otro país, constituirán registro sanitario por homologación en el Estado Plurinacional de Bolivia.
Lo mismo ocurre en el caso de Paraguay, donde mediante Resolución S.G. Nº 74653, establece:
Autorizar con carácter de emergencia las vacunas contra el COVID-19, autorizadas para uso de emergencia por las Autoridades Reguladoras de países de alta vigilancia sanitaria como FDA, EMA y ARNs de Referencia Regional.
Ejemplo de un reliance parcial, lo tenemos en Perú, que señala mediante Decreto Supremo 002-2021 S.A54:
(…) El plazo de evaluación de la solicitud es de hasta treinta días calendarios, cuando el producto se encuentre aprobado en la EMA o en los Países de Alta Vigilancia Sanitaria; o los mismos hayan sido precalificados por la OMS (…).
En cuanto al tiempo de evaluación empleado para este mecanismo (reliance) por parte de las ARNs, se tiene un valor medio de 15 días.50
Es importante destacar que las ARNs que recurren a las decisiones de otra siguen siendo responsables de las decisiones adoptadas y debe rendir cuenta de ellas.43
b. Autorización de Uso de emergencia
Con esta denominación se hace referencia a los procedimientos de evaluación de moléculas nuevas por parte de una ARN. Esta autorización es otorgada a partir del análisis de beneficio y riesgo de los datos limitados sobre la calidad, la seguridad y la eficacia disponibles a la fecha de autorización. Son procesos de evaluación continua que se realizan a medida que el laboratorio presenta la información.42
Este mecanismo se utiliza principalmente por las ARN de referencia regional, tal como se observa en la Tabla 2. Su empleo, permitió la inclusión de vacunas no listadas por la OMS, FDA o EMA. Como ejemplo, podemos ofrecer los casos de:
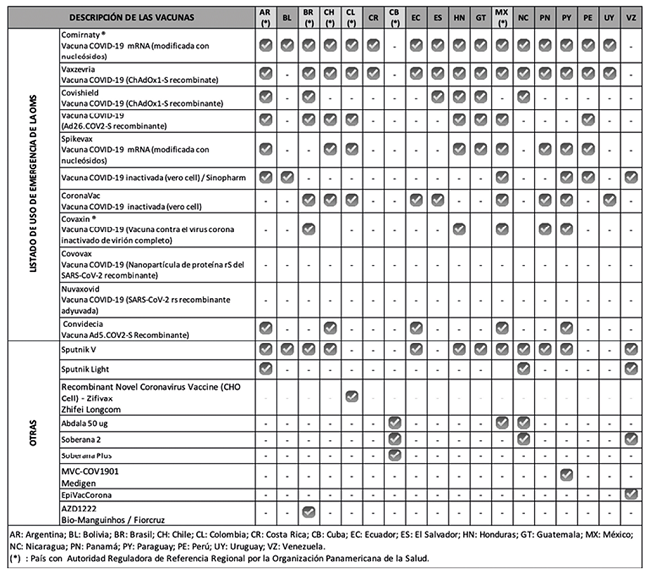
Argentina, ANMAT con fecha 23/12/2020 fue el primero en autorizar la Vacuna Sputnik V del Centro Nacional Gamaleya de Epidemiologia y Microbiología de Rusia.55 En base a esta autorización inicial, la utilización de la vacuna se extendió a Brasil, Bolivia, Chile, Ecuador, Honduras, México, Nicaragua, Panamá, Paraguay y Venezuela, como se observa en la Tabla 3.
Colombia, INVIMA con fecha 21/01/2022 otorgo la autorización a la vacuna Zifivax (la cual solo ha sido autorizado en Uberkistan, Pakistan, China e Indonesia).56
Cuba, CECMED autorizó la vacuna Abdala del Centro de Genética, Ingeniería y Biotecnología, y las vacunas Soberana 2 y Soberana Plus, ambas del Instituto Finlay. Esta autorización inicial por parte de una ARN de referencia, permitió el uso en México, Nicaragua y Venezuela, como se observa en la Tabla 3.
c. Registro en Condiciones Normales
En este supuesto se hace referencia a la obtención de una aprobación mediante la utilización de una vía convencional de registro. Es importante destacar que, de todos los mecanismos indicados, éste es el único que no implica una potencial reducción de requisitos o agilización de procesos.42
Ejemplo de la aplicación de este mecanismo lo tenemos en ANVISA (Brasil), que aprobó con carácter definitivo la vacuna de Pfizer el 23/02/2021.57 Esta vacuna, a diferencia de las aprobaciones de AstraZeneca (12/03/2021)58 y Janssen (5/04/2022)59, no paso por una autorización de emergencia previa. En América Latina, ANVISA es la única ARN en otorgar este tipo de autorización para las vacunas de COVID-19.
d. Procedimiento abreviado:
Este mecanismo hace alusión al uso de procedimientos simplificados, cuyo objetivo es agilizar la obtención de la autorización requerida para el medicamento.
Siguiendo a la OPS, en su análisis sobre la Preparación Regulatoria para la introducción de vacunas COVID-19 en la Región de las Américas42 podemos afirmar que la normativa relacionada a las autorizaciones de las vacunas para la COVID-19 no establece con claridad los requisitos y los tiempos aplicables que puedan diferenciar este mecanismo de los otros antes descritos.
Vacunas
Los procedimientos empleados por las distintas ARNs ponen de manifiesto la optimización del uso de sus recursos. La FDA y la EMA usaron procedimientos ya existentes en sus flujos estándares, tales como el pre-IND, y el asesoramiento científico y rolling review, respectivamente (todas ellas utilizadas en la evaluación de productos nuevos), los cuales fueron empleados para acelerar sus procesos de autorización de la EUA o la ACC. Esta optimización permitió que estas ARNs estuvieran entre las primeras en autorizar una vacuna para la COVID-19, y por tanto también fueron de las primeras en demandar dicho instrumento terapéutico.
Por su parte en el ámbito de América Latina, se hizo un uso extenso del reliance y de la autorización de uso de emergencia, con el fin de acceder en el menor tiempo posible al mayor número posible de vacunas, para dar una rápida respuesta a la falta de vacunas y a la demanda de sus programas de vacunación.
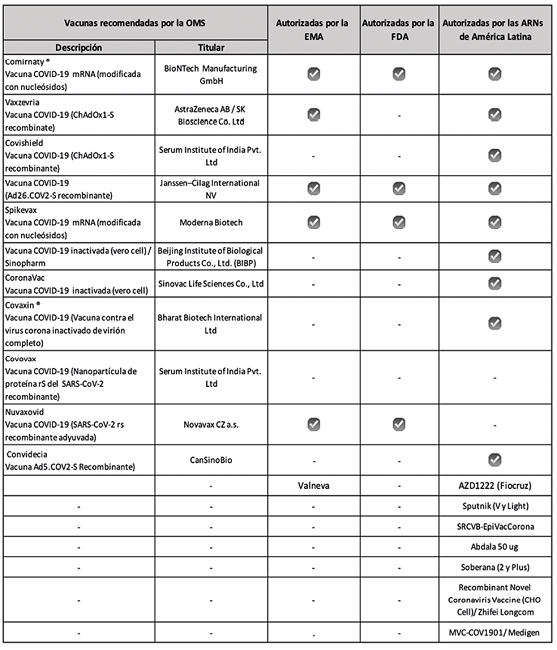
En definitiva, el uso de los distintos procedimientos aplicados por las ARNs, ofreció como resultado la aparición de una diversidad de vacunas utilizadas para la contención de la pandemia, tal como muestra la Tabla 4.
Conclusión
Los procedimientos usados por las ARNs, para autorizar el uso de las vacunas para la COVID-19 son diversos, pero todos ellos están orientados a proporcionar de manera rápida un producto de calidad, seguro y eficaz, que sirva como herramienta contención a la pandemia. El resultado de la utilización de esta diversidad de procedimientos, es la diversidad también de vacunas aprobadas en los EE.UU., la Unión Europea y América Latina.

