 ]]>
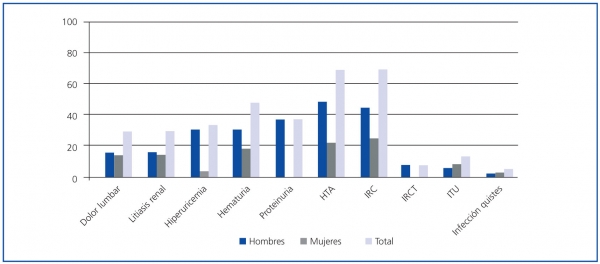
Figura 1. Manifestaciones renales de la enfermedad por sexo en el momento del diagnóstico
]]>
Figura 1. Manifestaciones renales de la enfermedad por sexo en el momento del diagnóstico
Análisis clínico de una población con poliquistosis renal autosómica dominante
Clinical analysis of a population with autosomal dominant polycystic kidney disease
Pilar Fraile Gómez, P. García-Cosmes, L. Corbacho Becerra, J.M. Tabernero Romo
Servicio de Nefrología. Hospital Universitario de Salamanca. Salamanca (España)
Dirección para correspondencia
]]>
RESUMEN
La poliquistosis renal autosómica dominante es una enfermedad hereditaria multiorgánica, responsable del 7-10% de los casos de insuficiencia renal crónica terminal que precisan tratamiento renal sustitutivo, causada por mutaciones en los genes PKD1 y PKD2. El diagnóstico de esta enfermedad puede realizarse fácilmente mediante pruebas radiológicas; la ecografía constituye el método de elección, pero el diagnóstico molecular ofrece la ventaja de la detección precoz de individuos asintomáticos portadores del defecto genético. En este trabajo, presentamos los resultados del análisis clínico de 48 pacientes diagnosticados de poliquistosis renal autosómica dominante. Los objetivos de nuestro trabajo fueron analizar los principales aspectos clínicos de la enfermedad, las causas de morbimortalidad e identificar a los individuos de riesgo afectados y sus manifestaciones clínicas precoces. En nuestro estudio, la hipertensión arterial fue la manifestación inicial más frecuente (68,42%), mientras que en la evolución de la enfermedad lo fue la insuficiencia renal crónica (100%). A pesar de que la edad media del diagnóstico de la poliquistosis renal en este estudio fue menor en las mujeres, la evolución de la enfermedad fue más tórpida en los hombres, lo que determinó el inicio más precoz del tratamiento renal sustitutivo y, consecuentemente, la mayor mortalidad. En este estudio se observó una prevalencia similar de muertes de origen cardiovascular (42,1%) e infeccioso (42,1%). En resumen, nuestros resultados revelan una alta prevalencia de pacientes con poliquistosis renal diagnosticados tardíamente, lo que podría explicar la elevada morbimortalidad. Dada la alta prevalencia de insuficiencia renal crónica e insuficiencia renal crónica terminal secundaria a poliquistosis renal en nuestro estudio, el diagnóstico precoz de la poliquistosis conllevaría un mejor pronóstico en relación con un seguimiento clínico más estricto. Por tanto, al ser la hipertensión arterial la manifestación clínica más frecuente en el momento del diagnóstico, debería incluirse esta entidad nosológica en todos los casos con hipertensión arterial de etiología no filiada y, por otra parte, las complicaciones infecciosas deberían ser un signo de alerta en todo paciente con poliquistosis renal autosómica dominante.
Palabras clave: Hipertensión arterial, Insuficiencia renal crónica, Insuficiencia renal crónica terminal, PKD1, PKD2, Poliquistosis renal autosómica dominante.
ABSTRACT
Autosomal dominant polycystic kidney disease is a multi-organic hereditary disorder. It is responsible for 7-10% of cases of end stage renal failure. It is caused by mutations in the genes PKD1 and PKD2. The diagnosis of this disease can be performed through ultrasounds, but the molecular diagnosis offers some advantages, such as the early detection of asymptomatic individuals who carry this genetic defect, in order to perform a preventive monitoring and genetic counselling. In this work, we present the results of the clinical analysis of 48 patients diagnosed with autosomal dominant polycystic kidney disease. The objectives of this work were to analyze the main clinical aspects of the disease. The average age of appearance of the first symptoms was 41.17 ± 13.41 years in women and 49.91 ± 12.52 years in men (p <0.05). Arterial hypertension was the first sign of the disease (68.42%), with more cases in men than in women (p <0.05), followed by chronic renal failure (68.29 %). The most common renal symptom during the evolution of the disease was chronic renal failure, which was present in all the patients of the study, followed by proteinuria (92.31%), end-stage renal failure (89.58%) and arterial hypertension (87.23%). In summary, our results reveal a high prevalence of patients with polycystic kidney disease who received a late diagnosis. This could possibly explain the high morbi-mortality associated to this condition. Given the high prevalence of chronic renal failure and end-stage renal failure secondary to polycystic kidney disease in our study, the early diagnostic of the disease would carry better pronostic in relation with a more strict clinical follow-up. The arterial hypertension was the most frequent clinical manifestation of the disease in our study by what this entity should be included in all the hypertense patients of unknown etiology and on the other hand, the infectious complications should be a sign of alert in every patient with polycystic kidney disease.
Key words: Arterial hypertension, Chronic renal failure, End-stage renal failure, PKD1, PKD2, Autosomal dominant polycystic kidney disease
Introducción
La poliquistosis renal autosómica dominante (PQRAD) es una enfermedad hereditaria multiorgánica, caracterizada por el progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional. Afecta aproximadamente a una de cada 1.000 personas y es responsable del 7-10% de los casos de insuficiencia renal crónica que precisan tratamiento renal sustitutivo1. La PQRAD está causada por mutaciones en el gen PKD1, responsable del 85-90% de los casos, en el gen PKD2, responsable del 10-15% de los casos y, posiblemente en un tercer gen, PKD3, que aún no ha sido identificado2-4.
]]> La PQRAD es una enfermedad multisistémica, con manifestaciones renales y extrarrenales derivadas de la formación de quistes renales y, en muchos casos, de quistes asintomáticos en el hígado y páncreas5. Las principales manifestaciones renales son dolor abdominal o lumbar, hematuria, hipertensión arterial (HTA) y, con menor frecuencia, infección urinaria, litiasis, insuficiencia renal o masa renal palpable6. Las principales manifestaciones extrarrenales son aneurismas cerebrales, quistes hepáticos, enfermedad valvular cardíaca, divertículos colónicos y hernias abdominales7,8.El diagnóstico de la PQRAD se establece habitualmente mediante técnicas de imagen; la ecografía renal constituye el método de elección en mayores de 30 años. En edades más tempranas deben emplearse la tomografía computarizada (TAC) o la resonancia magnética nuclear (RMN)9. En pacientes menores de 30 años, el diagnóstico de certeza lo ofrece el estudio genético, que proporciona información adicional en el consejo genético10,11.
En nuestro estudio nos planteamos analizar las características clínicas de todos los pacientes con PQRAD que precisaron atención médica por nuestro servicio durante el periodo 1994-2005.
Material y métodos
Población estudiada
Hemos realizado el estudio clínico de 48 pacientes con PQRAD que precisaron atención médica en el Servicio de Nefrología del Hospital Universitario de Salamanca durante el período 1994-2005. Los pacientes habían sido diagnosticados de PQRAD de acuerdo con criterios clínicos y radiológicos.
Seguimiento clínico
Se analizaron aspectos clínicos de 48 pacientes diagnosticados de PQRAD que precisaron intervención médica por parte de nuestro servicio a lo largo de 11 años (1994-2005) y se realizó seguimiento clínico de los mismos hasta marzo de 2008. La información de todos los pacientes se obtuvo de las historias clínicas. Los principales parámetros analizados fueron prevalencia y edad de aparición de la enfermedad, aspectos relacionados con las manifestaciones clínicas iniciales de la enfermedad, con las manifestaciones clínicas aparecidas en la evolución, con las manifestaciones extrarrenales, y con la supervivencia y morbimortalidad de los pacientes diagnosticados de PQRAD.
Método estadístico
]]> El estudio estadístico se realizó con el paquete informático SPSS® para Mac® OS X (versión 13.0). Las variables cualitativas se expresaron como porcentajes y las variables numéricas como media y desviación estándar (X ± DE).El método estadístico utilizado fue la prueba de la t de Student para contrastar hipótesis sobre medias en poblaciones con distribución normal. Cuando no cumplían el principio de normalidad se utilizó el test estadístico no paramétrico de Mann-Whitney. La relación entre las variables cualitativas de dos o más categorías se realizó por medio de proporciones mediante el test de la chi cuadrado.
Se consideraron, con independencia del análisis y del carácter de la variable analizada, valores de p <0,05 como probablemente significativos.
Resultados
Se estudiaron clínicamente 48 pacientes, 21 mujeres y 27 hombres, diagnosticados de PQRAD. La edad media de aparición de los primeros síntomas fue 46,07 ± 13,49 años, 41,17 ± 13,41 años en las mujeres y 49,91 ± 12,52 años en los hombres (p <0,05). El 41,18% de las mujeres tuvieron las primeras manifestaciones de la enfermedad en el intervalo de edad comprendido entre los 30 y 40 años, mientras que el diagnóstico en los hombres fue fundamentalmente entre los 50 y 60 años.
No se encontraron diferencias estadísticamente significativas en la prevalencia de la enfermedad según sexo.
Manifestaciones clínicas renales iniciales
Como manifestación aislada, la HTA fue la alteración más frecuente y fue el signo inicial de la enfermedad en el 68,42% de los pacientes, con predominio en los hombres respecto a las mujeres (p <0,05), seguida de la insuficiencia renal crónica (IRC), presente en el 68,29% de los pacientes con PQRAD (figura 1).
]]>
Figura 1. Manifestaciones renales de la enfermedad por sexo en el momento del diagnóstico
No se encontraron diferencias estadísticamente significativas en la prevalencia de las diferentes manifestaciones renales iniciales por razón del sexo, con excepción de la HTA, proteinuria, hiperuricemia y litiasis renal en las que la prevalencia fue mayor en los hombres (p <0,05) que en las mujeres. La cuantificación de la proteinuria en el momento del diagnóstico fue siempre inferior a 300 mg/24 h.
Manifestaciones clínicas renales en la evolución
La manifestación renal más frecuente en la evolución fue la IRC, presente en el 100% de los pacientes estudiados, seguida de la proteinuria (92,31%) y de la insuficiencia renal crónica terminal (IRCT) (89,58 %) (tabla 1). La HTA estuvo presente en el 87,23% de los pacientes enfermos, de forma estadísticamente significativa en el grupo de edad de 30-60 años (p <0,01). La prevalencia de las manifestaciones renales en la evolución no mostró diferencias significativas entre hombres y mujeres (tabla 1).
Tabla 1. Prevalencia, edad de aparición y tiempo transcurrido desde el diagnóstico de
la enfermedad hasta la aparición de las diferentes manifestaciones clínicas renales por sexo 
]]> Aunque la prevalencia de infecciones del tracto urinario fue superior en las mujeres, no se demostró significación estadística. Las enterobacterias fueron responsables del 94,12% de los episodios, y Escherichia coli fue el agente etiológico aislado con más frecuencia (70,59%).
La edad media de aparición de la IRC fue de 50,02 ± 11,21 años, y la de la IRCT de 54,74 ± 10,07 años, con independencia del sexo (p >0,05) (tabla 1). Los pacientes diagnosticados de IRC se encontraban preferentemente en el grupo de edad de 30-60 años (p <0,05), del mismo modo que los pacientes con IRCT, en quienes la diferencia no fue estadísticamente significativa. Cuando la enfermedad fue transmitida por el padre, la edad media de aparición de la IRC y de la IRCT fue menor (p <0,01).
El tiempo de evolución hacia la IRC y la IRCT (figura 2) desde el diagnóstico de la enfermedad fue menor en los hombres que en las mujeres, así como el tiempo transcurrido desde el diagnóstico de IRC hasta el inicio de tratamiento renal sustitutivo (p <0,05) (tabla 1 y figura 3). Los pacientes con HTA tenían más probabilidad de presentar IRC (p <0,05).

Figura 2. Curva de supervivencia renal de los pacientes con poliquistosis renal autosómica dominante
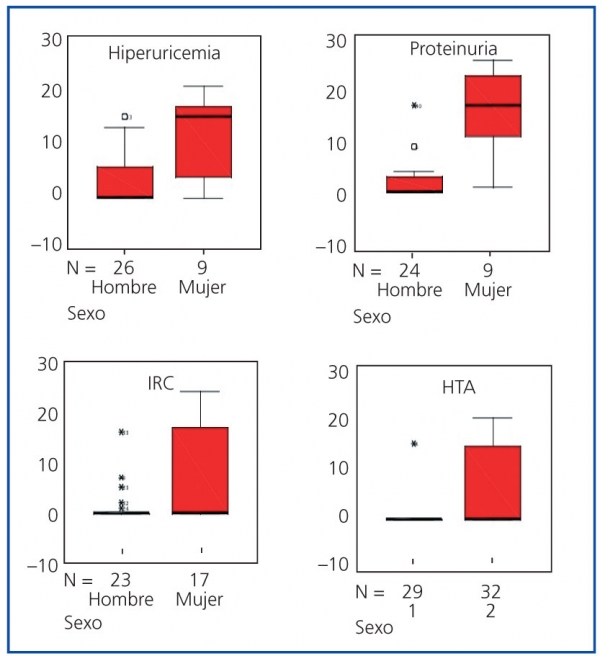
Figura 3. Distribución por sexos del tiempo transcurrido (años) desde el diagnóstico de la poliquistosis
renal autosómica dominante (PQRAD) hasta la aparición de hiperuricemia, proteinuria, IRC e HTA. ]]>
Se pone de manifiesto como el tiempo transcurrido desde el diagnóstico de la enfermedad hasta la
aparición de las manifestaciones clínicas descritas fue menor de forma estadísticamente significativa
en los hombres (p <0,05). Este tiempo fue también menor para el caso de la IRCT, cuyo box-plot
no aparece representado
Del total de pacientes que comenzaron tratamiento renal sustitutivo, el 83,72% eran hipertensos y el 13,95% normotensos. Los pacientes con HTA, proteinuria, hematuria macroscópica, litiasis, dolor abdominal, infecciones del tracto urinario, quistes hepáticos e infecciones de quistes tenían más probabilidad de presentar IRCT (p <0,01). Sin embargo, la probabilidad de presentar IRCT no aumentó en los pacientes con divertículos, hematuria macroscópica antes de los 30 años o mujeres con más de 3 hijos (p >0,05).
No se encontraron diferencias significativas por sexo en la edad de aparición de ninguna de las manifestaciones renales (tabla 1).
Además de en la IRC e IRCT, el tiempo de evolución desde el diagnóstico de la enfermedad hasta la aparición de hiperuricemia, proteinuria, litiasis renal e HTA fue también menor en los hombres que en las mujeres (p <0,05) (tabla 1 y figura 3). No se encontraron diferencias significativas en el tiempo de evolución transcurrido desde el diagnóstico de la enfermedad hasta la aparición del resto de las manifestaciones renales.
La litiasis renal recurrió de forma estadísticamente significativa en los hombres.
Manifestaciones extrarrenales
]]> Los quistes hepáticos fueron la manifestación extrarrenal más frecuente, presentes en el momento del diagnóstico en el 59,09% de los pacientes, con mayor prevalencia en mujeres (p <0,05) (figura 4). No se encontraron diferencias estadísticamente significativas en la prevalencia del resto de manifestaciones extrarrenales diagnosticadas por razón del sexo.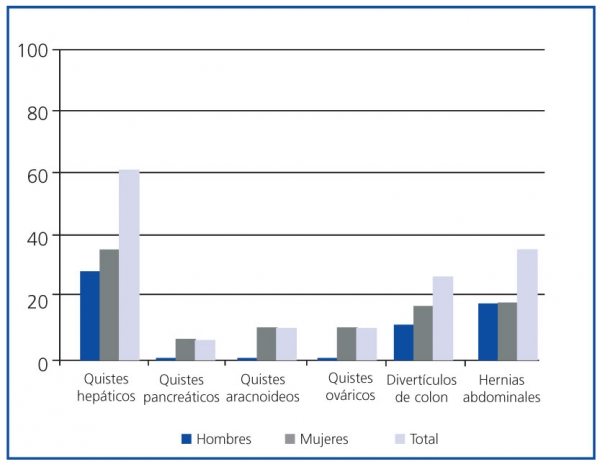
Figura 4. Manifestaciones extrarrenales de la poliquistosis renal autosómica dominante (PQRAD) por sexo
Supervivencia y morbimortalidad
Los pacientes con PQRAD presentaron un total de 6,17 ± 5,81 ingresos hospitalarios en nuestro centro hospitalario a lo largo de todo el estudio, 0,94 ± 0,63 ingresos/año, con una estancia media de 11,27 ± 4,91 días. No se encontraron diferencias significativas ni en la supervivencia, ni en el número de ingresos hospitalarios por año ni en la estancia media por razón del sexo (tabla 2).
Tabla 2. Prevalencia y significación estadística por sexo de los pacientes fallecidos, edad media en
el momento de la muerte, supervivencia desde el diagnóstico, ingresos hospitalarios y estancia media

]]> Del total de pacientes con PQRAD fallecieron durante el estudio el 39,58%, un 29,17% hombres y un 10,42% mujeres (p <0,05; RR = 2,3). La supervivencia de los pacientes desde el momento del diagnóstico fue de 13,4 ± 7,79 años, pero no se demostró que la HTA, la hematuria macroscópica, el dolor abdominal, las infecciones de quistes, la litiasis renal, las infecciones urinarias, la presencia de quistes hepáticos o la mayor paridad fueran factores de riesgo de mortalidad (p >0,05).
Las principales causas de fallecimiento fueron las de origen cardiovascular (42,1%) y las infecciosas (42,1%).
Discusión
Diagnóstico clínico
La PQRAD se diagnostica, en la mayoría de los pacientes, por las manifestaciones clínicas características, que suelen aparecer en la tercera o cuarta década de la vida. En nuestro estudio, la edad media de aparición de las primeras manifestaciones clínicas de la PQRAD fue menor en las mujeres que en los hombres. En los estudios publicados hasta el momento no se ha analizado la existencia de diferencias por razón del sexo en la edad del diagnóstico de la enfermedad o en la prevalencia de la mayoría de las manifestaciones renales y extrarrenales1,4,7,9,12.
En esta serie, la manifestación inicial más frecuente en el momento del diagnóstico de la enfermedad fue la HTA, presente en el 68,42% de los pacientes, con predominio en el sexo masculino y con una edad media de aparición (45,77 ± 10,48 años) más tardía que lo descrito en la literatura, con una media de 30 años13,14; no obstante, en nuestro estudio, precedió en varios años a los cambios medibles de la función renal1,13,15. La HTA es, según las diferentes publicaciones, uno de los signos guía para el diagnóstico de la PQRAD, dato confirmado en este estudio. El hecho de que el diagnóstico de la HTA en nuestra población fuera más tardío de lo publicado1,14,16 podría explicarse por la saturación actual de las consultas de medicina general y por la falta de realización de estudios protocolizados que aclaren la etiología de la HTA cuando ésta es leve o de fácil control; de este modo, se enmascararían casos de PQRAD en los que un tratamiento precoz y enérgico de las complicaciones retrasaría la aparición de IRC y la progresión a IRCT. Concuerda con esta hipótesis la alta prevalencia en nuestro estudio de IRC e IRCT secundaria a PQRAD como formas de presentación de la enfermedad. Durante la evolución de la PQRAD, la HTA estuvo presente en el 87,23% de nuestros pacientes, sin observar diferencias entre sexos. El tiempo de evolución desde el diagnóstico de la enfermedad hasta la aparición de HTA fue menor en los hombres. Se demostró su papel, ya descrito, en la progresión de la insuficiencia renal13,17, que no pudimos confirmar en menores de 35 años, dada la baja prevalencia de pacientes jóvenes en nuestro estudio. Tanto en la población general como en los enfermos con PQRAD se ha comprobado que la prevalencia de HTA es mayor en los hombres de forma significativa hasta los 45 años, momento a partir del cual aunque la prevalencia en éstos sigue siendo mayor, las diferencias dejan de ser significativas. El menor tiempo de evolución desde el diagnóstico de la enfermedad hasta la aparición de HTA en los hombres puede deberse a la existencia de factores de riesgo cardiovascular (hábito tabáquico, dislipemia, obesidad central) que favorecen el desarrollo de HTA y que predominan en los hombres con edades comprendidas entre 20 y 55 años. A partir de ese momento, a causa de la influencia hormonal, de la dislipemia o de la obesidad central, aumentan de manera significativa en las mujeres y, consecuentemente, lo hace la HTA14.
En esta serie, la proteinuria fue una de las manifestaciones renales más frecuentes de la PQRAD en el momento del diagnóstico, tras la HTA, la insuficiencia renal y la hematuria. Fue el signo de presentación de la enfermedad en el 36% de los hombres, de forma estadísticamente significativa, respecto a las mujeres, y el tiempo transcurrido desde el diagnóstico de la enfermedad hasta la aparición de la misma fue menor en los hombres. La HTA y la proteinuria presente en el momento del diagnóstico se han relacionado con peor pronóstico renal e incremento del riesgo cardiovascular en la PQRAD17,18. Dado que la HTA se correlaciona según las diferentes publicaciones con la aparición de proteinuria19, la mayor prevalencia de HTA en el momento del diagnóstico en los hombres del estudio explicaría también la mayor prevalencia de la proteinuria. Además, la HTA, la dislipemia, la obesidad troncular o el hábito tabáquico aumentan el riego de proteinuria y predominan en hombres en la edad media de la vida. Durante la evolución de la enfermedad la proteinuria pasó a ser la segunda manifestación renal más frecuente (92,31%), sin diferencias entre sexos, lo que se explicaría por el alto porcentaje de pacientes con daño renal establecido. La cuantificación de la proteinuria fue siempre inferior a 300 mg/dl, lo que concuerda con lo ya descrito19 y en ningún caso existieron proteinurias en rango nefrótico. En los pacientes con PQRAD estudiados con proteinuria se demostró que tenían más probabilidad de presentar IRCT, en relación con la hiperfiltración glomerular secundaria.
La hematuria estuvo presente, como síntoma de presentación de la enfermedad, en el 47,06% de los casos y, en la evolución de la enfermedad, en el 88,89% de nuestros pacientes con PQRAD. Esta prevalencia fue superior a la referida en la literatura, que la cifra en el 35-50%1. La hematuria puede deberse a cálculos renales, a infección de quistes o a tumores malignos. La elevada prevalencia en nuestra serie de litiasis renal e infecciones de quistes justificaría una prevalencia de la hematuria superior a la descrita. Se puso de manifiesto la asociación determinante de la hematuria macroscópica con la progresión de la insuficiencia renal17,20, pero no demostramos dicho papel en los pacientes menores de 30 años probablemente debido a la baja prevalencia de pacientes jóvenes con PQRAD. Este efecto deletéreo de la hematuria sobre la función renal puede explicarse tanto por los episodios obstructivos secundarios a la formación de coágulos como a las situaciones de hipovolemia que provoca.
La prevalencia de infecciones del tracto urinario en nuestro estudio fue similar a la descrita. Al igual que en la población general, por el motivo de que la uretra femenina es más corta, la prevalencia se elevó en mujeres, pero no de forma estadísticamente significativa12,16. E. coli fue el agente etiológico más frecuentemente aislado. Las infecciones más frecuentes fueron infecciones de quistes o pielonefritis agudas, con un papel determinante en la progresión de la insuficiencia renal, en probable relación con el deterioro de la función renal secundario a los episodios de inestabilidad hemodinámica derivados de infecciones graves.
]]> La IRC fue la segunda manifestación clínica más frecuente en el momento del diagnóstico y la de mayor prevalencia en la evolución de la enfermedad en nuestro estudio, mientras que en otras series estiman que sólo aproximadamente la mitad de los pacientes con mutación desarrollarán la enfermedad antes de la sexta década de la vida15. También las necesidades de tratamiento renal sustitutivo en nuestro trabajo fueron superiores a las descritas18. La edad media de aparición de la IRC (50,02 ± 11,21 años) y de la IRCT (54,74 ± 10,07 años) fue similar a la descrita en la literatura para pacientes con mutación en el gen PKD1 y necesidad de tratamiento renal sustitutivo (54,3 años)3,4, lo que nos induce a pensar en una mayor prevalencia de pacientes en nuestra población con mutación en el gen PKD1 si nos limitamos a realizar un análisis clínico. La edad de aparición de la IRCT fue menor cuando la enfermedad era transmitida por el padre que por la madre y el tiempo de evolución a IRC e IRCT desde el momento del diagnóstico de la enfermedad fue menor en los hombres que en las mujeres, dato ya reflejado en diferentes estudios3,21. En este estudio, los hombres tenían más probabilidad de progresar rápidamente a insuficiencia renal, lo que parece estar relacionado con un efecto directo de las hormonas sexuales21,22. También fue un dato de mal pronóstico en la evolución precoz tanto a IRC como a IRCT que la enfermedad fuera heredada del padre.En lo que se refiere a las manifestaciones extrarrenales8, éstas predominaron en las mujeres, aunque sólo se encontró significación estadística para los quistes hepáticos, cuya mayor prevalencia en mujeres se ha relacionado con el número de embarazos, la toma de anticonceptivos o la terapia hormonal sustitutiva. Aunque existen resultados contradictorios, confirmamos que las personas con quistes hepáticos presentan peor función renal que las que no los tienen3,16, probablemente a causa del efecto perjudicial que tendrían sobre la función renal las infecciones de repetición en los quistes hepáticos.
En este estudio se observó una prevalencia similar de muertes de origen cardiovascular (42,1%) e infeccioso (42,1%). Antes de 1975, la causa más frecuente de muerte en pacientes con PQRAD eran la infección (30%), la uremia (28%) y la enfermedad cardiovascular (21%). Con el desarrollo de la diálisis, la principal causa de mortalidad pasó a ser la cardiovascular (34%) seguido de las infecciones (20,4%)23. La principal causa de muerte de los pacientes en tratamiento renal sustitutivo y de los sometidos a trasplante renal es la cardiovascular; sin embargo, en nuestro estudio destaca la alta prevalencia de muertes de origen infeccioso, lo que se podría atribuir a la situación de inmunodeficiencia derivada de una población con una muy alta prevalencia de IRCT secundaria a PQRAD. La tasa de fallecimientos fue superior en los hombres, pero no se encontraron diferencias significativas en la supervivencia.
A pesar de que la edad media del diagnóstico de la PQRAD en este estudio fue menor en las mujeres, la evolución en los hombres hacia la aparición de litiasis renal, hiperuricemia, proteinuria, HTA, IRC e IRCT fue más corta. Esto, junto con la mayor prevalencia en hombres, en el momento del diagnóstico, de factores de riesgo involucrados en la progresión hacia la IRCT determinó una evolución más tórpida de la enfermedad en los hombres, el inicio más precoz del tratamiento renal sustitutivo y, consecuentemente, la mayor mortalidad. En las mujeres, sin embargo, el tiempo de evolución de la enfermedad fue mayor, lo que se tradujo en un mayor número de ingresos hospitalarios. Podríamos pensar que en las mujeres predomina la morbilidad y en los hombres la mortalidad, y probablemente no es reflejo de la enfermedad sino que es una observación común a toda la población.
En resumen, en nuestro estudio, los hombres portadores de PQRAD son diagnosticados más tardíamente que las mujeres, lo que podría explicar su peor evolución clínica. En esta población, la HTA fue la manifestación clínica más frecuente en el momento del diagnóstico, por lo que debería incluirse esta entidad nosológica en todos los casos con HTA no filiada, y las complicaciones infecciosas junto con las cardiovasculares fueron la principal causa de mortalidad, lo que implica que las complicaciones infecciosas sean un signo de alerta en todo paciente con PQRAD, incluidos los no sometidos a trasplante.
Referencias bibliográficas
1. Bleyer AJ, Hart TC, Wilson PD. Polycistic kidney disease. N Engl J Med 2004;350:2622. [ Links ]
2. Daoust MC, Reynolds DM, Bichet DG, Somlo S. Evidence for a third genetic locus for autosomal dominant polycystic kidney disease. Genomics 1995;25:733-6. [ Links ]
3. Brown JA. End stage autosomal dominant polycystic kidney disease. N Engl J Med 2002;347:1504. [ Links ]
4. Igarashi P, Somlo S. Polycystic kidney disease. J Am Soc Nephrol 2007;18:1371-3. [ Links ]
5. Romão EA, Moysés Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Braz J Med Biol Res 2006;39:533-8. [ Links ]
6. Demetriou K, Tziakouri C, Anninou K, Eleftheriou A, Koptides M, Nicolaou A, et al. Autosomal dominant polycystic kidney disease-type 2. Ultrasound, genetic and clinical correlations. Nephrol Dial Transplant 2000;15:205-11. [ Links ]
7. Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease. Am J Kidney Dis 1983;2:630-9. [ Links ]
8. Peces R, Venegas JL. Quistes de las vesículas seminales e infertilidad en la poliquistosis renal autosómica dominante. Nefrologia 2005;25:78-80. [ Links ]
9. Nicolau C, Torra R, Badenas C, Vilana R, Bianchi L, Gilabert R, et al. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology 1999;213:273-6. [ Links ]
10. Darnell A, Torra A, Badenas C, Pérez-Oller L, San Millán JL, Tellería D, et al. Estudio mutacional de los genes PKD1 y PKD2 (poliquistosis renal autosómica dominante). Nefrologia 2000;20:39-46. [ Links ]
11. Torres MJ, Rodríguez JC, Hernández CR, Anabitarte A, Caballero A, Vázquez C, et al. Diagnóstico molecular de la poliquistosis renal autosómica dominante en la Comunidad Autónoma de Canarias. Nefrologia 2006;26:666-72. [ Links ]
12. Torres VE. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Am J Kidney 1999;34:45-8. [ Links ]
13. Ecder T, Schrier RW. Hypertension in autosomal-dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001;12:194-200. [ Links ]
14. Kelleher CL, McFann KK, Johnson AM, Schrier RW. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease compared with general US population. Am J Hypertens 2004;17:1029-34. [ Links ]
15. Chapman AB. Autosomal dominant polycystic kidney disease: time for a change. J Am Soc Nephrol 2007;18:1399-407. [ Links ]
16. Gabow PA. Autosomal dominant polycystic kidney disease. More than a renal disease. Am J Kidney Disease 1990;16:403. [ Links ]
17. Choukroun G, Itakura Y, Albouza G, Christophe JL, Man NK, Grünfeld JP, et al. Factors influencing progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995;6:1634-42. [ Links ]
18. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007;369:1287-301. [ Links ]
19. Sharp CK, Gabow P. Factors relating to urinary protein excretion in children with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998;9:1908-14. [ Links ]
20. Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol 1997;8:1560-7. [ Links ]
21. Torra R, Badenas C, Darnell A, Nicolau C, Volpini V, Revert L, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol 1996;7:2142-51. [ Links ]
22. Burtey S, Lossi AM, Bayle J, Berland Y, Fontés M. Mutation screening of the PKD1 transcript by RT-PCR. J Med Genet 2002;39:422-9. [ Links ]
23. Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995;5:2048-56. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Pilar Fraile Gómez,
Servicio de Nefrología,
Hospital Universitario de Salamanca, Salamanca, España
E-mail: Pilarfg@usal.es