GFR: tasa de filtrado glomerular; HHRH: Raquitismo hipofosfatémico hereditario con hipercalciuria;
PTHi: paratohormona intacta; TP/GFR: Reabsorción tubular de fosfatos por 100 de GFR.
A]+[448+ 5G>A]). Sus tres hijos eran portadores de esta misma variante en heterocigosis y, aunque clínicamente estaban asintomáticos, dos de ellos tenían una hipercalciuria. Todos estos datos parecían indicar que el paciente presentaba un raquitismo hipofosfatémico hereditario con hipercalciuria (HHRH), secundario a una alteración en el cotransportador sodio-fosfato IIc (NaPi-IIc), localizado en el túbulo proximal. El HHRH se transmite de forma autosómica recesiva y es una forma muy rara de raquitismo hipofosfatémico. El diagnóstico y el tratamiento son fundamentales para evitar las secuelas óseas del raquitismo y la nefrocalcinosis. La distinción correcta con las otras formas de raquitismo hipofosfatémico tiene implicaciones en el tratamiento, ya que normalmente la administración aislada de suplementos de fósforo corrige todas las alteraciones clínicas y bioquímicas, excepto la pérdida de fosfato por la orina. El aporte exógeno de calcitriol, como se aconseja en otros raquitismos hipofosfatémicos, puede favorecer los depósitos renales de calcio y la aparición de nefrocalcinosis, así como empeorar su pronóstico.]]>
A] + [ 448 +5G>A] ). His three children were carriers of the same variant in heterozygosis and although they were clinically asymptomatic two of them had hypercalciuria. All these data suggest that the patient had hereditary hypophosphataemic rickets with hypercalciuria (HHRH) secondary to an alteration in the sodium dependent phosphate cotransporter located in proximal tubule (NaPi-IIc). The HHRH is transmitted by autosomal recessive inheritance and is an extremely rare form of hypophosphatemic rickets. The diagnosis and treatment are essential to prevent bone sequelae of rickets and nephrocalcinosis. A correct differential diagnosis with other forms of hypophosphatemic rickets has implications on the treatment, as the management based only on phosphorus supplementation usually corrects all clinical and biochemical abnormalities, except for the loss of phosphorus in the urine. The exogenous supply of calcitriol, as advised in other hypophosphatemic rickets, may induce renal calcium deposits and nephrocalcinosis and worsens the prognosis.]]>
Raquitismo hipofosfatémico hereditario con hipercalciuria: a propósito de un caso
Hereditary hypophosphatemic rickets with hypercalciuria: Case report
Ramón Areses-Trapote1, Juan A. López-García2, Mercedes Ubetagoyena-Arrieta1, Antxon Eizaguirre3, Raquel Sáez-Villaverde4
1Sección de Nefrología Pediátrica. Servicio de Pediatría. Hospital Universitario Donostia. San Sebastián-Donostia, Guipúzcoa
2Servicio de Urología. Hospital Universitario Donostia. San Sebastián-Donostia, Guipúzcoa
3Servicio de Nefrología. Hospital Universitario Donostia. San Sebastián-Donostia, Guipúzcoa ]]>
4Unidad de Genética. Hospital Universitario Donostia. San Sebastián-Donostia, Guipúzcoa
Dirección para correspondencia
RESUMEN
Presentamos el caso clínico de un varón de 50 años de edad que consulta por presentar una enfermedad renal litiásica recidivante y una nefrocalcinosis. En la exploración clínica destacó una talla baja y un genu varo bilateral importante. Entre los datos bioquímicos se apreciaba una pérdida renal de fosfatos intensa con hipofosfatemia, una 25 OH vitamina D3 normal, una 1,25 OH2 vitamina D3 elevada y una hipercalciuria. La hormona paratiroidea (PTHi) se encontraba disminuida y en la ecografía renal se confirmó la existencia de una nefrocalcinosis bilateral grave, localizada en la médula renal. Además, se constató una insuficiencia renal crónica incipiente y una acidosis tubular renal incompleta, ambas secundarias a la nefrocalcinosis y no directamente relacionadas con la enfermedad basal. En el estudio molecular se encontró un cambio en homocigosis en el intrón 5 del gen SLC34A3 (NM_080877.2:c[448+5G>A]+[448+ 5G>A]). Sus tres hijos eran portadores de esta misma variante en heterocigosis y, aunque clínicamente estaban asintomáticos, dos de ellos tenían una hipercalciuria. Todos estos datos parecían indicar que el paciente presentaba un raquitismo hipofosfatémico hereditario con hipercalciuria (HHRH), secundario a una alteración en el cotransportador sodio-fosfato IIc (NaPi-IIc), localizado en el túbulo proximal. El HHRH se transmite de forma autosómica recesiva y es una forma muy rara de raquitismo hipofosfatémico. El diagnóstico y el tratamiento son fundamentales para evitar las secuelas óseas del raquitismo y la nefrocalcinosis. La distinción correcta con las otras formas de raquitismo hipofosfatémico tiene implicaciones en el tratamiento, ya que normalmente la administración aislada de suplementos de fósforo corrige todas las alteraciones clínicas y bioquímicas, excepto la pérdida de fosfato por la orina. El aporte exógeno de calcitriol, como se aconseja en otros raquitismos hipofosfatémicos, puede favorecer los depósitos renales de calcio y la aparición de nefrocalcinosis, así como empeorar su pronóstico.
Palabras clave: Raquitismo/osteomalacia. Hipofostatemia. Hipercalciuria. Raquitismo hipofosfatémico. HHRH.
ABSTRACT
We report a case of a male aged 50 years who consulted for renal disease recurrent lithiasis and nephrocalcinosis. The clinical examination showed external signs of rickets/osteomalacia and biochemical data as well as a severe loss of renal phosphate with hypophosphatemia, normal 25 OH vitamin D, high 1,25 OH vitamin D and hypercalciuria. Parathyroid hormone was low and renal ultrasound confirmed the existence of severe bilateral medullary nephrocalcinosis. They also found incipient chronic renal failure and incomplete renal tubular acidosis, both secondary to nephrocalcinosis and unrelated to the underlying disease. The molecular study found a change in homozygosity in intron 5 of gene SLC34A3 (NM_080877.2:c[ 448 +5G>A] + [ 448 +5G>A] ). His three children were carriers of the same variant in heterozygosis and although they were clinically asymptomatic two of them had hypercalciuria. All these data suggest that the patient had hereditary hypophosphataemic rickets with hypercalciuria (HHRH) secondary to an alteration in the sodium dependent phosphate cotransporter located in proximal tubule (NaPi-IIc). The HHRH is transmitted by autosomal recessive inheritance and is an extremely rare form of hypophosphatemic rickets. The diagnosis and treatment are essential to prevent bone sequelae of rickets and nephrocalcinosis. A correct differential diagnosis with other forms of hypophosphatemic rickets has implications on the treatment, as the management based only on phosphorus supplementation usually corrects all clinical and biochemical abnormalities, except for the loss of phosphorus in the urine. The exogenous supply of calcitriol, as advised in other hypophosphatemic rickets, may induce renal calcium deposits and nephrocalcinosis and worsens the prognosis.
Key words: Rickets/osteomalacia. hypophosphatemia. hypercalciuria. Hypophosphatemic rickets. HHRH.
Introducción
Existen varias formas de raquitismo hereditario hipofosfatémico asociado a una disminución de la reabsorción renal de fósforo1-3. La mayoría de ellas se caracterizan por presentar una 1,25 OH2 vitamina D3 inapropiadamente normal o baja y una hipocalciuria, lo que indica que la anomalía subyacente que las produce altera el transporte renal de fósforo y la producción de calcitriol1-3.
La forma más conocida y prevalente es el raquitismo hipofosfatémico ligado al cromosoma X (XLRH), causado por mutaciones en el gen PHEX1-3.
Hace aproximadamente 30 años se describió una variante muy poco frecuente, conocida con el nombre de raquitismo hipofosfatémico hereditario con hipercalciuria (HHRH). Ésta, a diferencia de las otras formas de raquitismo hipofosfatémico, cursa con niveles elevados de 1,25 OH2 vitamina D3, hipercalciuria y una paratohormona (PTH) deprimida4-10.
En este estudio describimos un paciente varón adulto cuyas características fenotípicas y genéticas se corresponden con un HHRH.
Caso clínico
Presentamos el caso de un varón de 50 años de edad que fue valorado en el Servicio de Urología de nuestro hospital por presentar una enfermedad renal litiásica recidivante desde los 36 años de edad. En total, había formado 12 cálculos con expulsión espontánea del último cálculo unas semanas antes de acudir a nuestra unidad.
Antecedentes personales: Refiere que desde los primeros años de la vida presentaba una deformidad de las extremidades inferiores (EEII), con la pierna derecha arqueada hacia fuera y la izquierda hacia dentro, lo que le hacía deambular de una forma especial. Como no mejoraba, a los 13 años fue intervenido quirúrgicamente de ambas EEII, mejorando posteriormente la deambulación de forma aparente. En 1996 presentó una fractura múltiple en la extremidad inferior izquierda en relación con un accidente de coche, por lo que precisó varias intervenciones quirúrgicas. En la actualidad está pendiente de la colocación de una prótesis en la rodilla. Ha recibido tratamiento con calcio por vía oral durante muchos años. En los últimos meses le han detectado una hipertensión arterial que ha sido tratada primero con enalapril y posteriormente con amlodipino.
]]> Antecedentes familiares: El paciente tiene dos hermanos y tres hijos (dos varones y una mujer), y todos están sanos, sin presentar alteraciones óseas. Sus padres ya fallecieron, pero sabe que su padre tenía una enfermedad renal litiásica recidivante, por lo que fue intervenido en varias ocasiones.Exploración clínica: Peso: 67,2 kg. Talla: 148 cm. Baja estatura. Genu varo bilateral. El resto, sin hallazgos patológicos.
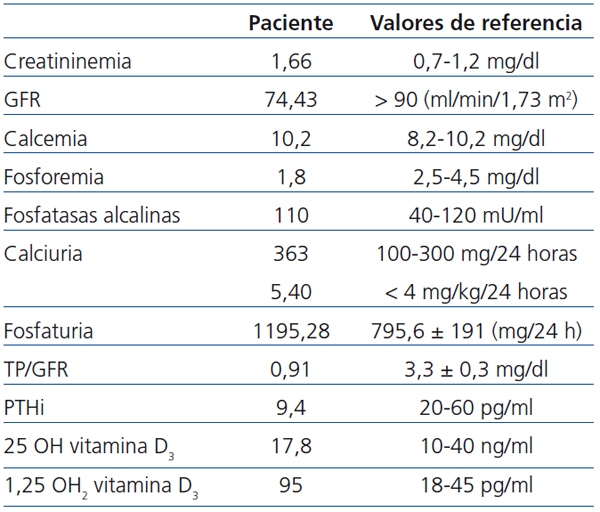
Exámenes complementarios: El estudio bioquímico sanguíneo basal realizado al paciente queda reflejado en la tabla 1. Además, presentaba una uremia de 55 mg/dl (valor de referencia [VR]: 10-50), un calcio iónico de 1,28 mmol/l (VR: 1,12-1,35) y una magnesemia de 1,8 mg/dl (VR: 1,59-2,5). En el estudio metabólico y de función renal realizado en orina de 24 horas presentaba: tasa de filtrado glomerular (GFR) discretamente disminuido de 74,43 ml/min/1,73 m2; una fosfaturia de 1195,28 mg/24 horas (VR: 795,6 ± 191); una reabsorción tubular de fosfatos (TRP) y una reabsorción tubular de fosfatos por 100 de GFR (TP/GFR) muy disminuidos, de 51% (VR: 87,44 ± 5,50) y de 0,91 mg/dl (VR: 3,3 ± 0,3), respectivamente; una hipercalciuria de 363 mg/dl (VR: 100-300), y una proteinuria leve glomerular < 0,3 g/24 horas. La β2-microglobulina y la α1-microglobulina en orina eran normales, lo que descartaba una proteinuria tubular. Los electrolitos séricos, el acido úrico y el oxalato eran normales tanto en sangre como en orina. Nunca se detectó glucosuria. El estudio de aminoácidos en sangre y orina, el aclaramiento y la reabsorción tubular de cada uno de ellos eran normales. La bicarbonatemia realizada en sangre capilar era normal. El pH urinario era alcalino de forma repetida, y la citraturia se encontraba en el límite inferior de lo normal, siendo de 399 mg/24 horas (VR: > 320 mg/24 horas).
Tabla 1. Parámetros bioquímicos del paciente con HHRH
GFR: tasa de filtrado glomerular; HHRH: Raquitismo hipofosfatémico hereditario con hipercalciuria;
PTHi: paratohormona intacta; TP/GFR: Reabsorción tubular de fosfatos por 100 de GFR.
Con el fin de descartar una acidosis tubular renal incompleta, se practicó un test de acidificación tras sobrecarga oral de cloruro amónico según el protocolo descrito previamente11. En el momento de máxima acidificación sanguínea, el pH urinario se mantuvo por encima de 5,5 (VR: 4,89 ± 0,24) y los niveles máximos de la acidez titulable, amonio y excreción neta de hidrogeniones, no alcanzaron los valores normales, siendo de 18,28 µEq/min/1,73 m2 (VR: 43,12 ± 10,21 ), 19 µEq/min/1,73 m2 (VR: 63,80 ± 19,54) y 36 µEq/min/1,73 m2 (VR: 107,17 ± 28,18), respectivamente (patrón analítico de acidosis tubular renal distal incompleta). Se practicó una sobrecarga oral cálcica después de siete días de una dieta exenta de calcio y sal, según el protocolo ya descrito12. Antes de la sobrecarga cálcica, estando en ayunas y tras la dieta hipocálcica, el cociente calcio/creatinina en orina de micción aislada era de 0,17 mg/mg. Tras la sobrecarga, dicho cociente se elevó por encima de lo normal, siendo de 0,23 mg/mg. La PTH intacta (PTHi) se mantuvo disminuida y la 1,25 OH vitamina D elevada (patrón analítico de hipercalciuria idiopática tipo III de Pak con absorción gastrointestinal de calcio aumentada y secundaria a una pérdida renal de fósforo). En el estudio radiológico realizado de forma repetida (radiografía simple de abdomen, ecografía renal y urografía intravenosa), presentaba una nefrocalcinosis medular bilateral muy importante.
]]> Estudio genético: En el estudio molecular realizado al paciente (reacción en cadena de la polimerasa y secuenciación bidireccional), se encontró un cambio en homocigosis en el intrón 5 del gen SLC34A3 (NM_080877.2:c.[448+5G>A]+[448+5G>A]). El análisis in silico no resolvió la benignidad o patogenicidad de la variante, por lo que se procedió a estudiar la segregación de ésta en sus tres hijos, mostrándose que los tres tenían el mismo cambio en el gen SLC34A3 en heterocigosis (portadores). No se ha podido realizar el estudio del ARN mensajero (ARNm). El estudio metabólico y de función renal de los tres hijos era normal, salvo que los dos varones presentaban únicamente una hipercalciuria leve13,14.
Discusión
El HHRH fue descrito por primera vez en 1985 por Tieder et al. en una familia beduina, exponiendo los casos de seis miembros afectados4. Se trata de un proceso muy poco frecuente que se transmite de forma autosómica recesiva y se manifiesta desde la niñez. Los pacientes con esta enfermedad presentan raquitismo, deformidades óseas, baja estatura, debilidad muscular y dolor óseo. El cuadro se caracteriza por un raquitismo/osteomalacia hipofosfatémico secundario a una exagerada pérdida renal de fosfato y a una elevación de la 1,25 OH2 vitamina D3 circulante, como respuesta a la hipofosfatemia. De forma secundaria aparece una hipercalciuria, debido a un aumento de la absorción gastrointestinal de fósforo y calcio, con depresión de la función paratiroidea. Son precisamente la hipercalciuria y los niveles elevados de 1,25 OH2 vitamina D3 los que diferencian el HHRH de otras formas de raquitismo/osteomalacia hipofosfatémico2,15,16. Como consecuencia de la hipercalciuria, se puede originar una nefrocalcinosis o litiasis, así como una insuficiencia renal.
Recientemente se ha publicado que el HHRH (OMIM#241530) se debe a mutaciones en el gen SLC34A3, localizado en el locus cromosómico 9q34 que codifica el cotransportador sodio-fosfato IIc (NaPi-IIc), que se expresa en la membrana apical de las células tubulares proximales renales y que normalmente regula la reabsorción del fosfato filtrado, bajo el control de la PTH y del factor de crecimiento fibroblástico (FGF 23). Las mutaciones con pérdida de función descritas en este gen son la causa de la excreción aumentada de fosfato a nivel proximal, característica de la enfermedad2,10,13 pudiendo dar lugar a diferentes cambios fenotípicos (figura 1)17,18.
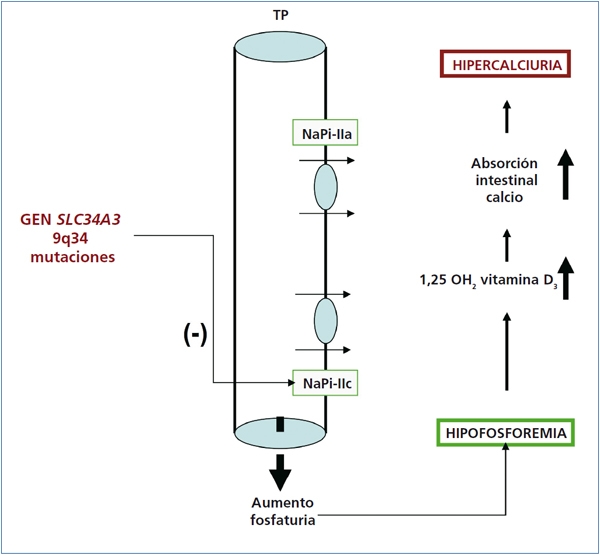
Figura 1. Representación esquemática de la reabsorción de fósforo en la membrana apical del túbulo
proximal y las consecuencias bioquímicas de las mutaciones con pérdida de función en el gen SLC34A3.
]]> En estudios realizados en pacientes con HHRH se ha descrito que simplemente con un suplemento de sales de fosfato por vía oral se pueden corregir todas las manifestaciones clínicas, analíticas y radiológicas características del cuadro, excepto la pérdida renal excesiva de fosfato. Ello sugiere que la hipofosfatemia, en ausencia de alteraciones en el metabolismo de la vitamina D, desempeña un importante papel en el metabolismo óseo6,8.
Por otro lado, puesto que la nefrocalcinosis probablemente está vinculada a la hipercalciuria, la administración de calcitriol, calcio o el incumplimiento del tratamiento con los suplementos de fósforo pueden favorecer los depósitos renales de calcio y ensombrecer el pronóstico en estos pacientes6,8.
Actualmente se considera que existe un eje hueso-riñón en el que están implicados varios genes y mediante el cual se regula el metabolismo del fósforo y la mineralización de la matriz ósea. El principal exponente, entre otros, de este eje es el FGF 23, la primera fosfatonina que fue descubierta y que es producida por los osteocitos. El NaPi-IIc, junto a otras proteínas como PHEX, MEPE y DMP1, participa en la regulación de este eje (tabla 2)2,3,19,20.
Tabla 2. Tipos de raquitismos hipofosfatémicos, genes implicados y características bioquímicas de cada uno de ellos 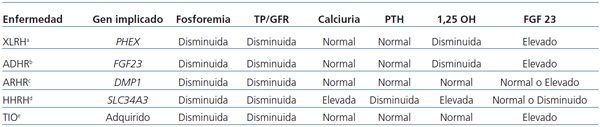
a Raquitismo hipofosfatémico ligado al cromosoma X.
b Raquitismo hipofosfatémico autosómico dominante.
c Raquitismo hipofosfatémico autosómico recesivo.
d Raquitismo hipofosfatémico hereditario con hipercalciuria. ]]>
e Raquitismo hipofosfatémico inducido por tumores.
TP/GFR: reabsorción tubular de fosfatos por 100 de GFR;
PTH: Paratohormona; GFR: tasa de filtrado glomerular
El paciente descrito en este trabajo reunía todas las características clínicas y analíticas compatibles con un HHRH. Al raquitismo/osteomalacia hipofosfatémico y a la reabsorción tubular de fosfato disminuida se a asociaban una elevación de la 1,25 OH2 vitamina D3 circulante y una hipercalciuria. Ello descartaba que se tratase de un XLRH, que es la forma más común de raquitismo hipofosfatémico familiar, que se debe a mutaciones inactivantes en el gen PHEX (OMIM#307800) y donde la PTH es normal y la 1,25 OH2 vitamina D3 anormalmente baja para los niveles de fosfatemia3,15,20. Por el mismo motivo, se podrían descartar también tres formas más de raquitismo hipofosfatémico: la hipofosfatemia tumor-inducida, que es secundaria a diversas fosfatoninas secretadas por tumores benignos de origen mesenquimal (sFRP-4, MEPE, FGF-7)20; el raquitismo hipofosfatémico autosómico dominante, que es una forma de raquitismo hipofosfatémico muy rara con penetrancia variable, que bioquímicamente es igual al XLRH y que se debe a mutaciones en el gen codificador del FGF 23 (OMIM#193100)21, y el raquitismo hipofosfatémico autosómico recesivo (OMIM#241520), causado por mutaciones en el gen DMP120. Por último, la ausencia de una proteinuria tubular descartaba también la enfermedad de Dent, caracterizada por ser una enfermedad hereditaria, transmitida de forma ligada al cromosoma X y causada por mutaciones en el gen CLCN5 que codifica el intercambiador electrogénico Cl-/H+, que pertenece a la familia de los canales/transportadores de iones cloruro, que se expresan en los endosomas del epitelio tubular proximal y que cursan también con hipercalciuria (tabla 2)22.
Nuestro paciente presentaba una acidosis tubular renal incompleta, reflejada claramente en el test de acidificación. Es conocida la existencia de una disfunción en la capacidad de acidificación renal en los pacientes con hipercalciuria y nefrocalcinosis, considerándose que esta última sería la causante de la acidosis. La insuficiencia renal presente en nuestro paciente igualmente era secundaria a la nefrocalcinosis, sin estar directamente relacionada con la propia enfermedad original23-25.
El estudio molecular constató un cambio en homocigosis en el intrón 5 del gen SLC34A3 (NM_080877.2:c[448+5G>A]+[448+ 5G>A]). El hecho de que los tres hijos sanos del paciente presentasen la misma variación de secuencia en el gen SLC34A3 en heterocigosis y que los dos varones presentaran una hipercalciuria iría a favor de que la alteración genética detectada fuera la causante del raquitismo hipofosfatémico. Recientemente se ha descrito una mutación localizada también en el intrón 5 (NM_080877.2:c.448+1G>A), muy próxima a la variante encontrada en nuestro paciente17.
En conclusión, presentamos un paciente con un raquitismo hipofosfatémico que, por los síntomas clínicos, los signos bioquímicos y el estudio genético, es compatible con HHRH. El diagnóstico y tratamiento precoz en estos pacientes es fundamental para evitar las secuelas óseas del raquitismo y la nefrocalcinosis. La distinción correcta con las otras formas de raquitismo hipofosfatémico tiene implicaciones en el tratamiento, ya que normalmente la administración aislada de suplementos de fósforo corrige todas las alteraciones clínicas y bioquímicas de la enfermedad, excepto la pérdida de fosforo por la orina. El aporte exógeno de calcitriol, como se aconseja en otros raquitismos hipofosfatémicos, puede favorecer los depósitos renales de calcio y la aparición de la nefrocalcinosis, así como empeorar su pronóstico.
Estudio molecular realizado en el Institut für Humangenetik des KlinikuMs rechts der Isar der Technischen Universität München. Direcktor: Univ. Prof. Med. Th. Meitinger POLIKLINIK.
]]> Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias Bibliográficas
1. Sermet-Gaudelus I, Garabédian M, Dechaux M, Lenoir G, Rey J, Tieder M. Hereditary hypophophatemic rickets with hypercalciuria: Report of a new kindred. Nephron 2001;88:83-6. [ Links ]
2. Negri AL. Hereditary hypophosphatemias: New genes in the bone-kidney axis. Nephrology 2007;12:317-20. [ Links ]
3. Shaikh A, Berndt T, Kumar R. Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol 2008;23:1203-10. [ Links ]
]]>4. Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, et al. Hereditary hypophophatemic rickets with hypercalciuria. N Engl J Med 1985;312:611-7. [ Links ]
5. Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, et al. Idiopathic hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect. N Engl J Med 1987;316:125-9. [ Links ]
6. Tieder M, Arie R, Bab I, Liberman UA. A new kindred with hereditary hypophosphatemic rickets with hypercalciuria: implications for correct diagnosis and treatment. Nephron 1992;62:176-81. [ Links ]
7. Nishiyama S, Inoue F, Matsudo I. A single case of hypophosphatemic rickets with hypercalciuria. J Pediatr Gastroenterol Nutr 1986;5:826-9. [ Links ]
8. Chen C, Carpenter T, Steg N, Baron R, Anast C. Hypercalciuric hypophosphatemic rickets, mineral balance, bone histomorphometry, and therapeutic implications of hypercalciuria. Pediatrics 1989;84:276-80. [ Links ]
]]>9. Navarro JF, Teruel JL, Montalban C, Gallego N, Ortuno J. Hypercalciuria secondary to chronic hypophosphatemia. Miner Electrolyte Metab 1994;20:255-8. [ Links ]
10. Mejia-Gaviria N, Gil-Peña H, Coto E, Pérez-Menéndez TM, Santos F. Genetic and clinical peculiarities in a new family with hereditary hypophosphatemic rickets with hypercalciuria: a case report. Orphanet J Rare Dis 2010;5:1. [ Links ]
11. Chahin J, García Nieto V, Torres A, Gallego E, Muros M, León C, et al. Defecto parcial de acidificación en pacientes con litiasis renal, con capacidad intacta de descender el pH urinario. Nefrologia 1993;XIII:556-60. [ Links ]
12. Pak CYC, Kaplan R, Bone H, Townsend J, Waters O. A simple test diagnosis of absortive, resorptive and renal hypercalciurias. N Engl J Med 1975;292:497-500. [ Links ]
13. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditay hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 2006;78:193-201. [ Links ]
]]>14. Bergwitz C, Roslin N, Tieder M, Loredo-Osti C, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodiumphosphate cotransporter NaPIIc in maintaining phosphate homeostasis. Am J Hum Genet 2006;78(2):179-92. [ Links ]
15. Santos F, Amil B, Chan JCM. Síndromes hipofosfatémicos. García Nieto V & Santos F (eds). Nefrología Pediátrica. 2.a ed. Madrid: Grupo Aula Médica; 2006. p. 161-79. [ Links ]
16. Yamamoto T, Michigami T, Aranami F, Segawa H, Yoh K, Nakajima S, et al. Hereditary hypophosphatemic rickets with hypercalciuria: a studhereditary hypophosphatemic rickets with hypercalciuria: a study for the phosphate transporter gene type IIc and osteoblastic function. J Bone Miner Metab 2007;25:407-13. [ Links ]
17. Phulwani P, Bergwitz C, Jaureguiberry G, Rasoulpour M, Estrada E. Hereditary hypophosphatemic rickets with hypercalciuria and nephrolithiasis-identification of a novel SLC34A3/NaPi-IIc mutation. Am J Med Genet A 2011;155A:626-33. [ Links ]
18. Tencza AL, Ichikawa S, Dang A, Kenagy D, McCarthy E, Econs MJ, et al. Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/Type IIc sodium-phosphate cotransporter: Presentatión as hypercalciuria and nephrolithiasis. J Clin Endocrinol Metab 2009;94:4433-8. [ Links ]
]]>19. Shiavi SC, Kurman R. The phosphatonin pathway: New insights in phosphate homeostasis. Kidney Int 2004;65:1-14. [ Links ]
20. Alon US. Fibroblast growth factor (FGF)23: a new hormone. Eur J Pediatr 2011;170:545-54. [ Links ]
21. The ADHR consortium. Autosomal dominant hypophosphatemic rickets is associated with mutations en FGF23. Nat Genet 2000;26:345-8. [ Links ]
22. Thakker RV. Phatogenesis of Dent´s, disease and related syndromes of X-linked nephrolithiasis. Kidney Int 2000;57:787-93. [ Links ]
23. García Nieto V, Monge M, Hernández L, Callejón A, Yanes MI, García VE. Estudio de la capacidad de acidificación renal en los niños diagnosticados de hipercalciuria idiopática. Nefrologia 2003;23:219-24. [ Links ]
]]>24. Rodríguez Soriano J, Vallo Boado A. renal tubular acidosis. Pediatr Nephrol 1990;4:268-75. [ Links ]
25. Seikaly M, Browne R, Baum M. Nephrocalcinosis associatrd with renal tubular acidosis in children with X-linked hypophosphatemia. Pediatrics 1996;97:91-3. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ramón Areses-Trapote
Sección de Nefrología Pediátrica. Servicio de Pediatría
Hospital Universitario Donostia, Po Beguiristain s/n. ]]>
20014 San Sebastián-Donostia, Guipúzcoa
ramon.aresestrapote@osakidetza.ner
rareses@hotmail.com