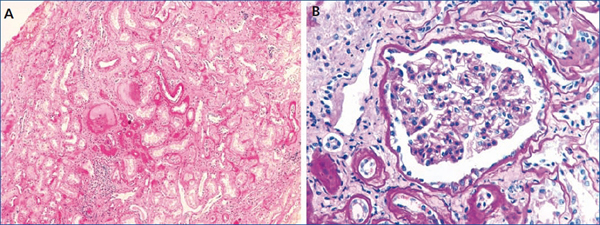
Figura 1. Biopsia renal del caso índice
A: Fibrosis intersticial, atrofia tubular focal, esclerosis glomerular y leve infiltrado inflamatorio
crónico (tinción H&E). B: Glomérulo sin alteraciones morfológicas ópticas (tinción PAS).
CASOS CLÍNICOS
Nefropatía intersticial crónica familiar con hiperuricemia causada por el gen UMOD
Familial chronic interstitial nephropathy with hyperuricaemia caused by the UMOD gene
Nadia Ayasreh-Fierro1, Elisabet Ars-Criach2, Vanesa Lopes-Martín3, Yolanda Arce-Terroba4, Patricia Ruiz-del Prado2, José Ballarín-Castán5, Roser Torra-Balcells1
1Enfermedades Renales Hereditarias. Servicio de Nefrología. Fundación Puigvert. Barcelona ]]>
2Laboratorio de Biología Molecular. Fundación Puigvert. Barcelona
3Servicio de Nefrología. Hospital Universitario Príncipe de Asturias, Universidad de Alcalá (REDINREN). Alcalá de Henares, Madrid
4Anatomía Patológica. Fundación Puigvert. Barcelona
5Servicio de Nefrología. Fundación Puigvert. Barcelona
Dirección para correspondencia
Introducción
Los avances en la genética médica en las últimas décadas han permitido mejorías en el diagnóstico de nefropatías familiares con gran variabilidad fenotípica que hasta ahora quedaban etiquetadas como nefropatías no filiadas. De manera global, estas nefropatías se pueden manifestar en forma de patología glomerular (sedimento de orina activo con proteinuria y/o hematuria) o bien en forma de nefropatía intersticial (hiperuricemia con o sin crisis gotosas, sedimento de orina no activo o escasamente activo, y frecuentemente clínica de poliuria y polidipsia), pero la gran variabilidad intrafamiliar hace que el diagnóstico resulte en muchas ocasiones dificultoso.
Desde el descubrimiento del gen UMOD1 y la reciente implementación de su estudio molecular en casos de nefropatía familiar con clínica intersticial y un patrón de herencia autosómica dominante, el número de pacientes identificados con mutaciones en este gen ha aumentado considerablemente.
]]> Se describe el caso de una familia con mutación del gen UMOD que presenta una gran variabilidad fenotípica intrafamiliar y en la que el estudio genético ha permitido el diagnóstico precoz de familiares jóvenes. Asimismo, se realiza una revisión de la literatura de esta entidad y de los casos descritos hasta la actualidad.
Caso clínico
El caso índice es un varón que inició el control en nuestro centro a la edad de 19 años por presentar un cuadro de mal estado general y cansancio desde hacía seis meses con alteración de la función renal (creatinina de 1,8 mg/dl). Presentaba hiperuricemia de cinco años de evolución sin episodios de crisis gotosa, que controlaba con alopurinol (100 mg/día). En la ecografía renovesical se observó disminución del tamaño de ambos riñones (9-10 cm) y en el sedimento de orina no había proteinuria ni hematuria. Se realizó una biopsia renal (figura 1), en la que se observan cambios inespecíficos: 4 glomérulos eran normales y 3 estaban en oblea, los túbulos tenían aspecto atrófico y existía un infiltrado linfomononuclear. Los vasos no estaban afectados y la inmunofluorescencia fue negativa.
Figura 1. Biopsia renal del caso índice
A: Fibrosis intersticial, atrofia tubular focal, esclerosis glomerular y leve infiltrado inflamatorio
crónico (tinción H&E). B: Glomérulo sin alteraciones morfológicas ópticas (tinción PAS).
]]> El paciente presentó una insuficiencia renal de lenta progresión y a los 39 años se realizó un trasplante renal de donante vivo (su esposa) sin incidencias. Actualmente, a los 42 años de edad, presenta una función renal muy estable con creatininas basales de 1,5-1,7 mg/dl y está en tratamiento con alopurinol 100 mg/día.
En los antecedentes familiares constan varios miembros con afectación renal e hiperuricemia, con diferentes grados de insuficiencia renal y clínica de poliuria y polidipsia (figura 2) (tabla 1). Un hermano (III-2) con enfermedad renal crónica e hiperuricemia, trasplantado a los 47 años. Otra hermana (III-1), también con enfermedad renal crónica e hiperuricemia (a los 35 años consta una creatinina de 1,8 mg/dl). La madre (II-4) de los tres hermanos, también con enfermedad renal crónica, inició diálisis a los 34 años y falleció a los 66 años.
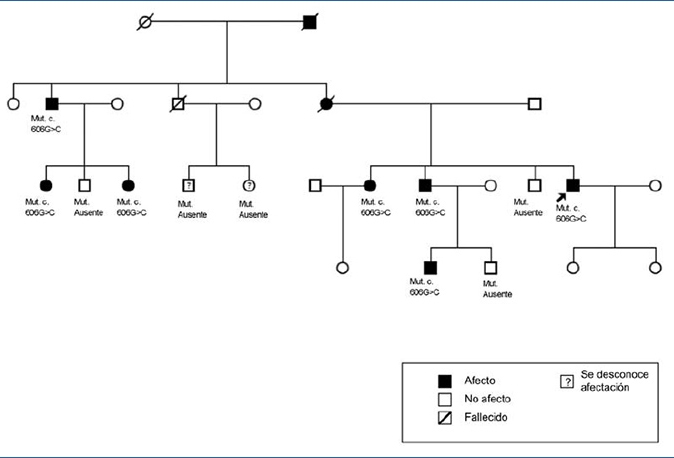
Figura 2. Pedigrí de la familia.
Se indica el caso índice y los familiares estudiados con y sin mutación.
Tabla 1. Características clínicas de los familiares afectados 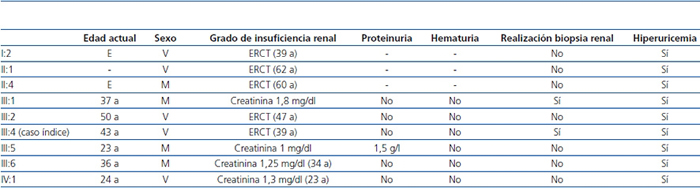
a: años; E: exitus; ERCT: enfermedad renal crónica terminal; M: mujer; V: varón.
El diagnóstico de nefropatía hiperuricémica familiar juvenil se realizó mediante el análisis mutacional del gen UMOD mediante secuenciación directa de los 10 exones codificantes (exón 2-exón 11) a partir del ADN genómico del caso índice. El análisis mutacional permitió la identificación de la variante de secuencia c.606G>C (p.W202C) en heterocigosis (figura 3). Esta variante se localiza en el exón 3 del gen UMOD y no ha sido previamente descrita en la bibliografía, aunque ha sido descrita otra mutación que altera el mismo aminoácido [c.605G>C (p.W202S)2]. La variante c.606G>C (p.W202C) no se identificó en más de 200 cromosomas controles analizados y altera el aminoácido triptófano 202 que está totalmente conservado en las proteínas ortólogas. Además, se estudió la segregación de esta variante en la familia y se identificó que está compartida por todos los miembros afectados. Distintos algoritmos bioinformáticos (Condel, Sift, Polyphen) predijeron que se trata de una mutación patogénica. Por todo ello, concluimos que la variante c.606G>C (p.W202C) era con muy elevada probabilidad la mutación patogénica causante de la enfermedad.

Figura 3. Fragmento de secuencia del exón 3 del gen
UMOD
A: mutación en el caso índice (p.W202C). B: secuencia control.
El diagnóstico de esta mutación patogénica permitió el estudio presintomático de algunos de los familiares jóvenes, como es el caso de los individuos IV:1 (afecto) y IV:2 (no afecto).
Discusión
]]> La uromodulina (UMOD), o proteína de Tamm-Horsfall (640 aminoácidos, peso molecular de 85-90 kDa)1, expresada en el asa ascendente de Henle, es la proteína más abundante en la orina en condiciones normales. Su función, aunque no ha sido del todo definida, se ha relacionado con la impermeabilización del túbulo distal, un efecto protector de infecciones del tracto urinario y litiasis renales, así como con una actividad proinflamatoria3,4. La disfunción de esta proteína se ha relacionado con la incapacidad de concentrar la orina y la fibrosis tubulointersticial3. En los pacientes con mutación de UMOD se observa una disminución en la concentración de esta proteína en la orina por un plegamiento anormal que produciría su acumulación en las células del epitelio tubular5. Aunque las patologías causadas por mutación del gen UMOD no se incluyen generalmente en la clasificación de las ciliopatías6, se han publicado en la literatura algunos estudios que sí demostrarían expresión de UMOD en el cilio primario7.Las mutaciones en el gen UMOD (región cromosómica 16p2)8,9 son responsables de dos nefropatías tubulointersticiales con patrón de herencia autosómica dominante, la enfermedad medular quística tipo 2 (MCKD2) [OMIM 603860] y la hiperuricemia familiar juvenil [OMIM 162000], que actualmente se engloban en la denominada «enfermedad renal causada por mutación en el gen UMOD»5,10-14. Un gen localizado en el locus 1q21 MCKD1 [OMIM 174000], pero no identificado hasta la fecha15-18, es responsable también de nefropatía tubulointersticial con patrón de herencia autosómica dominante, con fenotipo semejante, pero con una presentación más precoz.
La enfermedad renal causada por mutación en el gen UMOD tiene una prevalencia muy baja, menos del 1% de los casos de enfermedad renal terminal (ERT) en edad adulta, aunque puede que esté infradiagnosticada5. En nuestra serie de casos de nefropatías familiares con clínica de nefropatía intersticial hiperuricémica, se ha detectado mutación en el gen UMOD en un 12,5% de ellas (datos no publicados), en consonancia con los publicados en la literatura19.
La deficiencia de la uromodulina causa un déficit de reabsorción tubular, favoreciendo la poliuria y la hiperuricemia13. La fracción excretada de ácido úrico aparece disminuida de forma precoz (incluso en pacientes jóvenes con función renal todavía normal), y suele ser menor del 5% en varones adultos y menor del 6% en mujeres adultas20. Se han observado niveles de ácido úrico por encima del percentil 75 en más del 70% de estos pacientes. Aproximadamente el 75% de los varones y el 50% de las mujeres presentan crisis gotosas19, pero también se han descrito casos con niveles de ácido úrico en sangre dentro de la normalidad, sobre todo en mujeres21. Cabe destacar la desproporción de la hiperuricemia respecto al grado de insuficiencia renal19. Todos los miembros de la familia presentada tenían hiperuricemia desde la juventud y no constaban antecedentes de crisis gotosas.
La insuficiencia renal es lentamente progresiva, llegando por lo general a la fase terminal entre la cuarta y la sexta década de la vida19. El sedimento de orina es no productivo (en fases de ERT puede aparecer proteinuria habitualmente menor de 1 g/día) y en la ecografía renal suelen observarse riñones de tamaño disminuido y en algunas ocasiones pequeños quistes medulares (en algunas series, se ha observado en una tercera parte de los pacientes19). Los grados de enfermedad renal y las edades de presentación son muy variables tanto a nivel intrafamiliar como interfamiliar13,19,22-24. En nuestro caso todos los familiares afectados presentaron hiperuricemia y, a excepción del individuo III:5, tuvieron un sedimento de orina normal. El individuo III:5 presentó, en algún sedimento de orina de seguimiento, proteinuria leve, 1-1,5 mg/dl. Aunque en la literatura la mayoría de los casos descritos cursan sin proteinuria, existe algún caso con proteinuria leve25. Cabe destacar la gran variabilidad intrafamiliar en cuanto a la evolución de la enfermedad. La edad de ERT oscila desde formas precoces (edad aproximada de 40 años), como el caso índice o los individuos I:2 y III:2, a formas tardías (edad aproximada de 60 años), como en el caso de los individuos II:1 o II:4. Por ello, el diagnóstico de estas nefropatías puede resultar difícil si la carga familiar no es muy importante. Los casos familiares con ligamiento para MCKD1 acostumbran a tener un mayor grado de hiperuricemia y ataques de gota más frecuentes y precoces, con una llegada a fases finales de la insuficiencia renal más precoz16,26,27.
A nivel histológico, puede observarse nefritis crónica intersticial, atrofia tubular y fibrosis intersticial, y en ocasiones infiltrado linfocítico. La lesión principal es el adelgazamiento y la pérdida progresiva de la membrana basal tubular y la formación de quistes en el túbulo distal y tubos colectores. En la inmunohistoquímica pueden verse depósitos anormales de uromodulina en las células tubulares11,28. En el presente caso, la biopsia renal se realizó solo en 2 de los 9 casos afectados. En el caso índice (individuo III:4) se realizó en el momento del diagnóstico de la enfermedad (a la edad de 20 años), 20 años antes de llegar a ERT, y se observaron cambios inespecíficos. En el individuo III:1 también se realizó biopsia renal de forma precoz (a la edad de 29 años), observando pequeños focos de atrofia tubular y ectasia microquística de túbulos aislados, con una microscopía electrónica normal.
El estudio genético se basa en la secuenciación del gen UMOD y la detección de mutaciones (hasta la actualidad se han descrito unas 50 mutaciones)3,14,19,27. Dado que la mayoría de las mutaciones se localizan en los exones 3 y 4 del gen, estos suelen estudiarse en primer lugar en el caso índice, aunque también se han descrito mutaciones en otros exones, como el exón 719. No se ha observado una correlación clara entre fenotipo-genotipo13,19,29. El 90% de las mutaciones son de tipo cambio de aminoácido (missense) y el 62% alteran un residuo de cisteína, produciendo alteraciones en el plegamiento de la proteína30. En caso de ausencia de mutación en el gen UMOD, si se dispone de muestras de otros familiares afectados y no afectados, se puede realizar un análisis de ligamiento para la región MCKD18,17,23.
El gen UMOD está regulado por varios factores de transcripción, entre otros HNF1b, cuya mutación es responsable de otra enfermedad hiperuricemiante que se debería sospechar en casos de clínica compatible y los estudios anteriormente citados negativos7,31,32.
Las indicaciones primordiales del diagnóstico genético son el estudio de donante vivo, la posibilidad de ofrecer opciones reproductivas seguras y el diagnóstico presintomático. La mutación hallada en este caso no ha sido descrita previamente. Se trata de una mutación de tipo missense en un aminoácido muy conservado entre especies y con una correcta segregación familiar. El hallazgo de esta mutación permitió el diagnóstico presintomático del individuo IV:1 y descartar la enfermedad en el individuo IV:2.
No existe un tratamiento específico para esta enfermedad, así como para el resto de las enfermedades quísticas renales hereditarias5. El tratamiento con antihiperuricemiantes puede resultar de utilidad para reducir la progresión de la enfermedad renal33, aunque los resultados son muy variables. El tratamiento con inhibidores del eje renina-angiotensina está recomendado por su efecto nefroprotector, sin que existan tampoco ensayos que demuestren beneficios concretos para esta enfermedad. En el momento actual no hay ensayos de fármacos específicos para el tratamiento de la enfermedad renal asociada a la uromodulina.
]]>Conclusiones
La enfermedad renal causada por mutación en el gen UMOD es una entidad poco conocida y por su gran variabilidad intra e interfamiliar está seguramente muy infradiagnosticada. Se debería sospechar esta nefropatía en casos de enfermedad renal no filiada con patrón de herencia autosómica dominante, sedimento de orina normal y biopsia renal con fibrosis intersticial predominante. También hay que tener en cuenta que, al igual que en todas las enfermedades renales hereditarias, se tiende a estudiar a nivel genético a los pacientes con un fenotipo clásico, por lo que puede que el espectro fenotípico de la enfermedad sea mucho más amplio de lo descrito hasta la fecha.
Es probable que los avances en la genética médica permitan en un futuro no muy lejano identificar el gen MCKD1, así como otros genes implicados en nefropatía intersticial familiar. Por tanto, es de esperar que se pueda profundizar en el conocimiento de las nefropatías intersticiales familiares redundando en el hallazgo de posibles dianas terapéuticas que algún día faciliten el tratamiento de esta entidad.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias bibliográficas
1. Pennica D, Kohr WJ, Kuang WJ, Glaister D, Aggarwal BB, Chen EY, et al. Identification of human uromodulin as the Tamm-Horsfall urinary glycoprotein. Science 1987;236(4797):83-8. [ Links ]
2. Kudo E, Kamatani N, Tezuka O, Taniguchi A, Yamanaka H, Yabe S, et al. Familial juvenile hyperuricemic nephropathy: detection of mutations in the uromodulin gene in five Japanese families. Kidney Int 2004;65(5):1589-97. [ Links ]
3. Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int 2011;80(4):338-47. [ Links ]
4. El-Achkar TM, Wu XR. Uromodulin in kidney injury: an instigator, bystander, or protector? Am J Kidney Dis 2012;59(3):452-61. [ Links ]
5. Coto García E. Enfermedad renal quística medular y nefronoptisis. Nefrologia 2011;2(1):74-9. [ Links ]
6. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011;364(16):1533-43. [ Links ]
7. Zaucke F, Boehnlein JM, Steffens S, Polishchuk RS, Rampoldi L, Fischer A, et al. Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Hum Mol Genet 2010;19(10):1985-97. [ Links ]
8. Bleyer AJ. Improving the recognition of hereditary interstitial kidney disease. J Am Soc Nephrol 2009;20(1):11-3. [ Links ]
9. Hateboer N, Gumbs C, Teare MD, Coles GA, Griffiths D, Ravine D, et al. Confirmation of a gene locus for medullary cystic kidney disease (MCKD2) on chromosome 16p12. Kidney Int 2001;60(4):1233-9. [ Links ]
10. Hummel A. Familial juvenile hyperuricemic nephropathy. Nephrol Ther 2012;8(2):117-25. [ Links ]
11. Dahan K, Fuchshuber A, Adamis S, Smaers M, Kroiss S, Loute G, et al. Familial juvenile hyperuricemic nephropathy and autosomal dominant medullary cystic kidney disease type 2: two facets of the same disease? J Am Soc Nephrol 2001;12(11):2348-57. [ Links ]
12. Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 2002;39(12):882-92. [ Links ]
13. Scolari F, Caridi G, Rampoldi L, Tardanico R, Izzi C, Pirulli D, et al. Uromodulin storage diseases: clinical aspects and mechanisms. Am J Kidney Dis 2004;44(6):987-99. [ Links ]
14. Lens XM, Banet JF, Outeda P, Barrio-Lucia V. A novel pattern of mutation in uromodulin disorders: autosomal dominant medullary cystic kidney disease type 2, familial juvenile hyperuricemic nephropathy, and autosomal dominant glomerulocystic kidney disease. Am J Kidney Dis 2005;46(1):52-7. [ Links ]
15. Christodoulou K, Tsingis M, Stavrou C, Eleftheriou A, Papapavlou P, Patsalis PC, et al. Chromosome 1 localization of a gene for autosomal dominant medullary cystic kidney disease. Hum Mol Genet 1998;7(5):905-11. [ Links ]
16. Scolari F, Puzzer D, Amoroso A, Caridi G, Ghiggeri GM, Maiorca R, et al. Identification of a new locus for medullary cystic disease, on chromosome 16p12. Am J Hum Genet 1999;64(6):1655-60. [ Links ]
17. Hodanova K, Majewski J, Kublova M, Vyletal P, Kalbacova M, Stiburkova B, et al. Mapping of a new candidate locus for uromodulin-associated kidney disease (UAKD) to chromosome 1q41. Kidney Int 2005;68(4):1472-82. [ Links ]
18. Kirby A, Gnirke A, Jaffe D, Baresova V, Pochet N, Blumenstiel B, et al. Combination of modern and traditional techniques identify MCKD1 casual frameshift variants within the MUC1 VNTR. J Am Soc Nephrol 2012 (abstract). [ Links ]
19. Bollee G, Dahan K, Flamant M, Moriniere V, Pawtowski A, Heidet L, et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol 2011;6(10):2429-38. [ Links ]
20. Moro F, Ogg CS, Simmonds HA, Cameron JS, Chantler C, McBride MB, et al. Familial juvenile gouty nephropathy with renal urate hypoexcretion preceding renal disease. Clin Nephrol 1991;35(6):263-9. [ Links ]
21. Bleyer AJ, Hart TC. Familial juvenile hyperuricaemic nephropathy. QJM 2003;96(11):867-8. [ Links ]
22. Bleyer AJ. Improving the recognition of hereditary interstitial kidney disease. J Am Soc Nephrol 2009;20(1):11-3. [ Links ]
23. Smith GD, Robinson C, Stewart AP, Edwards EL, Karet HI, Norden AG, et al. Characterization of a recurrent in-frame UMOD indel mutation causing late-onset autosomal dominant end-stage renal failure. Clin J Am Soc Nephrol 2011;6(12):2766-74. [ Links ]
24. Lhotta K, Piret SE, Kramar R, Thakker RV, Sunder-Plassmann G, Kotanko P. Epidemiology of uromodulin-associated kidney disease - results from a nation-wide survey. Nephron Extra 2012;2(1):147-58. [ Links ]
25. Bleyer AJ, Hart PS, Kmoch S. Hereditary interstitial kidney disease. Semin Nephrol 2010;30(4):366-73. [ Links ]
26. Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I, et al. Autosomal-dominant medullary cystic kidney disease type 1: clinical and molecular findings in six large Cypriot families. Kidney Int 2002;62(4):1385-94. [ Links ]
27. Wolf MT, Mucha BE, Hennies HC, Attanasio M, Panther F, Zalewski I, et al. Medullary cystic kidney disease type 1: mutational analysis in 37 genes based on haplotype sharing. Hum Genet 2006;119(6):649-58. [ Links ]
28. Dahan K, Devuyst O, Smaers M, Vertommen D, Loute G, Poux JM, et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol 2003;14(11):2883-93. [ Links ]
29. Lhotta K, Piret SE, Kramar R, Thakker RV, Sunder-Plassmann G, Kotanko P. Epidemiology of uromodulin-associated kidney disease - results from a nation-wide survey. Nephron Extra 2012;2(1):147-58. [ Links ]
30. Williams SE, Reed AA, Galvanovskis J, Antignac C, Goodship T, Karet FE, et al. Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum Mol Genet 2009;18(16):2963-74. [ Links ]
31. Faguer S, Decramer S, Chassaing N, Bellanne-Chantelot C, Calvas P, Beaufils S, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 2011;80(7):768-76. [ Links ]
32. Wolf MT, Hoskins BE, Beck BB, Hoppe B, Tasic V, Otto EA, et al. Mutation analysis of the Uromodulin gene in 96 individuals with urinary tract anomalies (CAKUT). Pediatr Nephrol 2009;24(1):55-60. [ Links ]
33. Fairbanks LD, Cameron JS, Venkat-Raman G, Rigden SP, Rees L, Van'T HW, et al. Early treatment with allopurinol in familial juvenile hyerpuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease. QJM 2002;95(9):597-607. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Roser Torra-Balcells,
Enfermedades Renales Hereditarias. ]]>
Servicio de Nefrología, Fundación Puigvert,
08025, Barcelona
nayasreh@fundacio-puigvert.es
rtorra@fundacio-puigvert.es
Enviado a Revisar: 6 Feb. 2013
Aceptado el: 15 Abr. 2013