Genetic diagnosis in clinical psychiatry: A case report of a woman with a 47, XXX karyotype and Fragile X syndrome
Dr. Anthony M. Vandersteen*, David Moore**, Celia Donaghue***, Dr. Neil MacFarlane****, Dr. Dragana Josifova*
* Guy’s and St. Thomas’ NHS Foundation Trust, Dept. Clinical Genetics.
** Guy’s and St. Thomas’ NHS Foundation Trust, Dept. Molecular Genetics.
*** Guy’s and St. Thomas’ NHS Foundation Trust, Dept. Cytogenetics.
**** Consultant in Adult Developmental Psychiatry, Honorary Senior Lecturer, University of Kent. ]]>
ABSTRACT
Background and Objectives: A recent report highlighted the importance of considering a chromosomal abnormality in the differential diagnosis of adult clinical psychiatry. This case report illustrates the importance of considering Fragile X syndrome, an X-linked genetic disorder associated with psychiatric morbidities.
Methods: A 45 years old woman was referred to the clinical genetics department by her psychiatrist for investigation of her gross obesity, hyperphagia, learning difficulties and affective disorder.
Results: Cytogenetic analysis revealed a 47,XXX karyotype. Molecular testing identified an expansion of approximately 580 repeats in the FRAXA gene carried on two of her three copies of the X chromosome. Clinical evaluation revealed features consistent with the Prader-Willi like phenotype of Fragile X syndrome.
Conclusions: It is important to consider molecular and cytogenetic testing in patients with dysmorphic features, complex neuro-behavioural profile and/or psychotic disorders in order to establish a causative diagnosis, provide adequate counselling and initiate cascade screening where applicable.
Key words: Fragile X; 47,XXX; obesity; hyperphagia; affective disorder.
]]> Introduction
A recent report highlighted the importance of considering a genetic syndrome in the differential diagnosis of adults with learning difficulties, autism and psychiatric illness1. Most adults with learning difficulties have not had specific genetic investigations, which may reflect previously lower availability of testing as well as cultural and historical divides between healthcare professionals and those involved in the daily care of individuals with learning difficulties2.
Fragile X syndrome (FXS) is the most common inherited cause of learning difficulties in men. FXS is caused by expansion (>200 repeats) in a CGG repeat sequence of the FRAXA gene (Xq27.3). Female pre-mutation carriers (55-200 repeats) are at risk of premature ovarian failure and males of tremor/ataxia syndrome (FXTAS) later in life3.
FXS is associated with autism spectrum, anxiety and mood disorders, hyperactivity and aggressive behaviours in men3. Women with FXS may have anxiety, depression, learning and attention disorders even in the presence of normal or above average IQ scores4. A Prader-Willi like phenotype has been reported for men with FXS with no evidence of chromosome 15q abnormality5. We present the case of a 45 years old woman referred to the clinical genetics department for investigation of her gross obesity, hyperphagia, learning difficulties and affective disorder. This combination was consistent with a diagnosis of Prader-Willi Syndrome and she was referred to the Genetics Services for assessment and testing.
Case Report
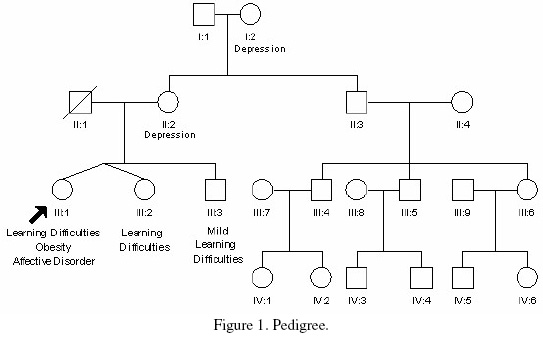
The patient is the third child of unrelated parents and is one of non-identical twins (Figure 1). Her mother gave no history of illness or substance misuse during pregnancy. There was no history of neonatal hypotonia or feeding difficulties, although irritability in infancy was reported. She achieved her motor milestones within the expected time, but presented with speech delay, communication and learning difficulties.
At age of 17 the patient underwent a formal assessment which revealed a full scale IQ of 44. A more recent assessment revealed a full scale IQ of 65 with a particular deficit in verbal reasoning. She never achieved independence and lived in a sheltered accommodation under supervision. She was known to have severe hyperphagia with nocturnal food foraging. She manifested an increasingly sexualised behaviour which was becoming of concern to the social services. At the age of 41 she developed a psychotic illness with a severe affective component and was under regular psychiatric review. She responded well to treatment.
]]> The proband’s twin sister and older brother were also known to have learning difficulties. They were not available for assessment in our clinic, however, they were known to be of similar stature and build, although not to the same extent as our patient. The patient’s brother had a history of psychiatric illness. Unfortunately we were unable to obtain more detail about their condition. The patient’s mother had a history of depression but was of normal cognitive ability, in good general health with no history of premature menopause.The patient was tall, with a height of 178cm (98th-99.6th centile), weight of 191Kg (>99.8th centile, BMI = 60) and a head circumference of 56cm (50th-75th centile). She had small hands and feet (shoe size 4) with normal skull shape, normal ear morphology and no specific dysmorphic features (Figure 2).

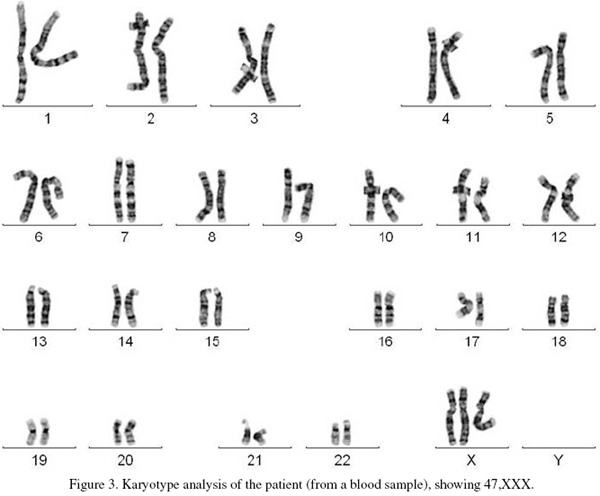
Cytogenetic analysis involved standard G-banded karyotype analysis (Figure 3). This showed a 47,XXX karyotype (Triple X), the result was confirmed by multiplex ligation-dependent probe amplification (MLPA). Quantitative fluorescence Polymerase Chain Reaction analysis (QF-PCR) of four informative markers on the X chromosome (DXS7423, HPRT, DXS1187 and DXYS287) were consistent with three copies of the X chromosome showing a 2:1 ratio for the maternal to paternal alleles.
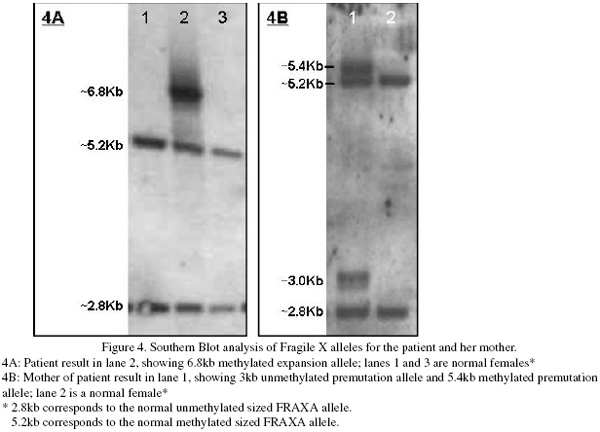
Patient DNA was isolated from EDTA anticoagulated peripheral blood. Amplification of the repeat regions at the FRAXA locus was carried out using standard fluorescent PCR amplification. A single FRAXA allele corresponding to 33 CCG repeats was amplified for the index case. Southern blot analysis showed an additional discrete band was present corresponding to a fully methylated, expanded allele of approximately 580 repeats (Figure 4) and was significantly darker than the normal sized bands. Laboratory testing of the patient’s mother’s blood sample showed amplification of a normal FRAXA allele corresponding to 34 repeats and a pre-mutation allele of approximately 84-92 repeats was detected. Unmethylated and methylated alleles were present in approximately equal ratios. FRAXE analysis showed normal sized alleles for the index case and her mother. Unfortunately no samples were available from the father of the index case.
The above investigations concluded that the index case carried three copies of the X chromosome, two of which were maternally inherited, both carrying a full expansion at the FRAXA locus.
Discussion
]]> To our knowledge an increased association of Fragile X syndrome and Triple X has not been reported. A single case report of a woman with Triple X who was found to carry a single copy of an X chromosome with an expanded FRAXA allele and two normal copies of the X chromosome was mentally and physically normal6.Nowicki et al.5 described the Prader-Willi like phenotype in males affected with Fragile X Syndrome: severe behavioural problems and mental retardation, onset of hyperphagia between the ages of 1 and 10, extreme obesity and short stature. Small hands and feet were reported previously in an original case series7,8.
Our patient presented with features of the Prader-Willi-like phenotype of Fragile X Syndrome including severe cognitive difficulties, behavioural problems, and morbid obesity secondary to extreme food consumption. The patient’s tall stature may be a feature of both Fragile X and Triple X Syndrome, although we can not exclude the possibility that there was a significant familial component in this case. Our patient had a relatively small head circumference, a feature more in keeping with Triple X than with Fragile X syndrome. The latter is more likely to be associated with macrocephaly9. To our knowledge the Prader-Willi Syndrome like phenotype in females with Fragile X has not been previously reported in the literature.
In females, X-inactivation takes place early in embryogenesis to provide dosage compensation. The X-inactivation is usually random resulting in equal representation of maternal and paternal genes on the X-chromosome. However, skewed X-inactivation is a well recognised mechanism that may produce a clinical phenotype of a traditionally X-linked recessive disorder in carrier women to the extent that they may present with a clinical picture very similar to that seen in affected males10. In our patient, with an assumption of random X-inactivation, one would expect a probability 2 out of 3 of each cell line carrying a FRAXA mutation as opposed to 1 out of 2 in female carriers with a normal chromosome complement. It is likely therefore that this dosage effect is the cause of this phenotype.
Conclusions
This case report highlights the importance of consideration of cytogenetic and molecular investigations in patients with a complex neuro-behavioural profile and/or psychotic disorders, particularly in the presence of a family history of learning difficulties and/or dysmorphic features. It is important to compile a detailed family history and consider genetic investigations in order to establish a causative diagnosis. Although this may not necessarily alter patient’s management it may prove crucial for other family members at risk to have the opportunity to receive appropriate counselling, take part in cascade screening and make informed choice where applicable.
References
1. Verhoeven WMA, Tuerlings JHAM, van Ravenswaay-Arts CMA, Boermans JAJ, Tuinier S. Chromosomal Abnormalities in Clinical Psychiatry a report of two older patients. Eur J Psychiat 2007; 21(3): 207-211. [ Links ]
2. Finucane B, Haas-Givler B, Simon E. Genetics, mental retardation, and the forging of new alliances. Am J Med Genet Part C: Seminars in Medical Genetics 2003; 117C(1): 66-72. [ Links ]
3. Garber KB, Visootsack J, Warren ST. Practical Genetics - Fragile X Syndrome. Eur J Hum Genet 2008; 16: 666-672. [ Links ]
4. Angkustsiri K, Wirojanan J, Deprey LJ, Gane LW, Hagerman RJ. Fragile X Syndrome with anxiety Disorder and Exceptional Verbal Intelligence. Am J Med Genet Part A 2008; 146A: 376-379. [ Links ]
5. Nowicki ST, Tassone, F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, et al. The Prader-Willi Phenotype of Fragile X Syndrome. J Dev Behav Pediatr 2007; 28(2): 133-138. [ Links ]
6. Fuster C, Templado C, Miró R, Barrios L, Egozcue J. Concurrence of the triple-X syndrome and expression of the fragile site Xq27.3. Hum Genet 1988; 78: 293. [ Links ]
7. de Vries BB, Fryns JP, Butler MG, Canziani F, Wesby-van Swaay E, van Hemel JO, et al. Clinical and molecular studies in fragile X patients with a Prader-Willi-like phenotype. J Med Genet 1993; 30(9): 761-766. [ Links ]
8. de Vries BB, Robinson H, Stolte-Dijkstra I, Tjon Pian Gi CV, Dijkstra PF, van Doorn J, et al. General overgrowth in the fragile X syndrome: variability in the phenotypic expression of the FMR1 gene mutation. J Med Genet 1995; 32 (10): 764-769. [ Links ]
9. Linden M. 47,XXX. In: Firth H.V. and Hurst JA editors. Oxford desk Reference Clinical Genetics: Oxford University Press; 2005. p. 494-495. [ Links ]
10. Migeon BR. The Role of X Inactivation and Cellular Mosaicism in Women’s Health and Sex-Specific Diseases. J Am Med Assoc 2006; 295: 1428-1433. [ Links ]
]]>
![]() Address for correspondence:
Address for correspondence:
Dr. Dragana Josifova
Dept of Clinical Genetics
Guy’s and St. Thomas’ NHS Foundation Trust
Great Maze Pond
London SE1 9RT
Fax: +44-207-188-1369
Tel +44-207-188-1364
E-mail: Dragana.josifova@gstt.nhs.uk.