
Large chondroma of the dural convexity in a patient with Noonan's syndrome. Case report and review of the literature
Condroma gigante de la convexidad en un paciente con síndrome de Noonan. Caso clínico y revisión de la literatura
P.D. Delgado-López; V. Martín-Velasco; A.Mª. Galacho-Harriero; J.M. Castilla-Díez; A. Rodríguez-Salazar y *C. Echevarría-Iturbe
Servicios de Neurocirugía y de *Anatomía Patológica. Hospital General Yagüe. Burgos.
Dirección para correspondencia
Introduction. Intracranial chondromas are extremely rare intracranial tumours that usually arise from the skull base synchondrosis. Exceptionally, they may grow from cartilage rests within the dura mater of the convexity or the falx. They may be part of Ollier's multiple enchondromatosis or Maffuci's syndrome. We describe the case of a young male diagnosed of Noonan's syndrome that underwent resection of a large intracranial chondroma arising from the dural convexity.To our best knowledge this is the first report of suchassociation.
Case report. An 18-year-old male presented with a single generalized seizure. The patient was previously diagnosed of Noonan's syndrome on the basis of his special phenotype (Turner-like), low stature, cardiac malformation, retarded sexual and bone development and normal karyotype. He harboured mild psychomotor retardation. Physical and neurological examinations were unremarkable. Brain Magnetic Resonance image showed a large well-circumscribed intracranial mass in the dural convexity of the left frontal-parietal lobes, with heterogeneous contrast enhancement and no peritumoural oedema. The patient was initiated on valproic acid and underwent craniotomy and complete excision of the tumour. The tumour was firm, white-greyish, avascular and could be finely dissected away from the cortex. Postoperative seizures required additional anticonvulsant therapy. He was discharged uneventfully. The pathological study revealed a mature chondroma. Subsequent brain MRI studies have shown no evidence of recurrence after 33 months of follow up.
Discussión. Chondromas comprise less than 0.3% of intracranial tumours. Only twenty-five cases of intracranial dural convexity chondromas are reported in the literature. Several hystopathogenetic theories have been proposed: metaplasia of meningeal fibroblasts and perivascular meningeal tissue, traumatic or inflammatory cartilaginous activation of fibroblasts and growth of aberrant embryonal cartilaginous rests in the dura mater. Chondromas present clinical features similar to meningiomas. CT scan imaging shows a mass of variable density due to different degrees of calcification with minimum to moderate contrast enhancement. MRI studies show a well-circumscribed lesion without surrounding tissue oedema, that exhibit heterogeneous signal with intermediate to low intensity on T1-weighted images and mixed intensity on T2-weighted images with minimum enhancement. Angiogram is clue to differentiate from meningiomas since chondromas are completely avascular. Complete tumour resection including its dural attachment is the treatment of choice. Long-term prognosis is favourable. Radiation therapy is currently not recommended for residual tumours or inoperable patients due to risk of malignization. Noonan's syndrome (also known as pseudo-Turner syndrome) is a complex familial genetic disorder with a phenotype that resembles that of Turner's syndrome but exhibits no chromosomal defect. No predisposition of Noonan's syndrome for tumoural development is reported in the literature. Association of a dural convexity chondroma with Noonan's syndrome is unique as far as the literature is concerned.
Key words: Chondroma. Noonan's syndrome. Epilepsy. Tumer's syndrome.
RESUMEN
Introducción. Los condromas intracraneales son tumores extremadamente raros que suelen surgir de la sincondrosis de la base craneal. Excepcionalmente, puede crecer a partir de restos cartilaginosos en la duramadre de la convexidad o en la hoz. Se han relacionado con la encondromatosis múltiple de Ollier y con el síndrome de Maffuci. Describimos el caso de un varón joven diagnosticado de síndrome de Noonan en el que se resecó un condroma gigante de la convexidad. Esta asociación no está descrita en la literatura hasta el momento.
Caso clínico. Varón de 18 años de edad que presenta una única crisis comicial generalizada como debut clínico. Estaba previamente diagnosticado de síndrome de Noonan basándose en su fenotipo especial (Turner-like), baja estatura, presencia de malformación cardíaca, retraso en la maduración ósea y sexual, y cariotipo normal. No presentaba alteraciones significativas en la exploración física y neurológica salvo un leve retraso mental. El estudio de resonancia magnética cerebral mostró una masa intracraneal de gran tamaño, bien circunscrita, dependiente de la convexidad dural frontoparietal izquierda, con captación heterogénea de contraste y sin edema perilesional. Comenzó tratamiento con ácido valproico y se realizó una resección completa de la lesión. El tumor era de consistencia dura, blanco-grisáceo, avascular y pudo disecarse por completo de la corteza. Presentó crisis comiciales postoperatorias que precisaron tratamiento combinado con un segundo anticomicial. Anatomía patológica: condroma maduro. Las RM de control han mostrado ausencia de recidiva tras 33 meses de seguimiento.
Discusión. Los condromas comprenden menos del 0,3% de los tumores intracraneales. Hasta la fecha, sólo se han descrito veinticinco casos de condromas de convexidad dural en la literatura. Se han propuesto diversas teorías histopatogénicas: metaplasia de fibroblastos meníngeos y tejido meníngeo perivascular, activación traumática o inflamatoria de fibroblastos hacia cartílago, y crecimiento de restos cartilaginosos embrionarios aberrantes en la duramadre. Los condromas presentan características clínicas similares a los meningiomas. La imagen de TAC muestra una masa de densidad variable debido a los diferentes grados de calcificación con mínima a moderada captación de contraste. Los estudios de RM evidencian una masa bien circunscrita sin edema perilesional, de señal heterogénea, hipointensa en T1 y de intensidad mixta en T2, y con captación mínima de contraste. La angiografía los diferencia perfectamente de los meningiomas pues aquéllos son totalmente avasculares. El tratamiento de elección es la resección completa incluyendo la duramadre adyacente. El pronóstico a largo plazo es excelente. El tratamiento con radioterapia no se recomienda ni en los restos tumorales ni en los pacientes inoperables, debido al riesgo de malignización. El síndrome de Noonan (también conocido como pseudo-Turner) es una enfermedad genética familiar compleja cuyo fenotipo se asemeja al del síndrome de Turner pero no presenta defecto cromosómico. Hasta la fecha, no se ha descrito en la literatura una predisposición al desarrollo de tumores en los pacientes con Noonan ni tampoco la asociación de este síndrome con un condroma de convexidad cerebral.
Palabras clave: Condroma. Síndrome de Noonan. Epilepsia. Síndrome de Turner.
Introduction
Intracranial chondromas are very rare tumours with an estimated incidence of 0.2 to 0.3% of all intracranial tumours3,16. Most of these neoplasms arise from a synchondrosis located at the skull base, especially in the sellar and parasellar areas13,15. Exceptionally, they may grow from cartilage rests within the dura mater of the convexity or the falx3,4,10,14.
These tumours are typically diagnosed in young adults and exhibit no clear sex predominance3,16. They may present as raised intracranial pressure, focal neurological disturbances, cranial nerve palsies, hydrocephalus or seizures13. Intracranial chondromas are considered hystologically benign lesions, sometimes associated to certain syndromes such as Ollier's multiple enchondromatosis or Maffuci's syndrome8,19. A mixed tumour formed by mature cartilage and choroid plexus papilloma has been also reported21.
We describe the case of a young male previously diagnosed of Noonan's syndrome that underwent craniotomy and resection of a large intracranial chondroma arising from the dural convexity. To our best knowledge this is the first report of such association. The literature concerning intracranial dural chondromas is reviewed.
Case report
An 18-year-old male presented with a single generalised seizure which occurred during sleep. The patient was previously diagnosed of Noonan's syndrome on the basis of his special phenotype: short neck, hypertelorism, broad nasal base, anti-mongoloid ocular obliquity, fishlike mouth, ojival soft palate, cubitus valgus, angel's wing, retarded sexual characters development, low stature and normal karyotype (46 XY). He was born slightly premature (caesarean section at 34 weeks, weighing 2650 grams) and suffered hydramnios and neonatal sepsis. Two older brothers had died at ages 3 and 16 days postpartum respectively, due to complex cardiac malformations. A patient's niece was also diagnosed of Noonan's syndrome and one of his aunts had epilepsy of unknown origin. He was diagnosed of mild pulmonary valve stenosis. He had an abnormal bone development (corresponding to fourteen years-old) and his stature had been below percentile 3 ever since birth.
At the time of admission he was 154 cm tall, weighed 50.9 Kg and he was in Tanner stage III-IV of sexual development. His parents referred mild psychomotor retardation enough as to interfere with normal schooling.
Complete physical and neurological examinations were unremarkable. Laboratory studies detected a mild coagulation defect (Prothrombine time between 60 and 72%; INR: 1.32) but no other abnormality. Echocardiography showed the pulmonary valve stenosis, and hormone values and blood biochemical parameters were normal. A fusion defect of the spinal posterior arch was found at the L5 level. Brain Magnetic Resonance image showed a large well-circumscribed intracranial mass apparently extending from the dural convexity of the left frontal-parietal lobes, with heterogeneous contrast enhancement, no peritumoural oedema and a remarkable space-occupying effect (figure 1). The patient was initiated on valproic acid and he was prepared for surgery after the appropriate informed consent was obtained.
]]> The patient underwent left frontal-parietal craniotomy and complete excision of the tumour. It was infiltrative in the dura mater and eroded the inner table of the skull. It showed a striking lobulated appearance mimicking cortical gyri and sulci (figure 2). A defined arachnoid plane allowed fine dissection from the underlying cortex as in classicalconvexitymeningiomas.Thetumourwasfirm,whitegreyish, avascular and too difficult to aspirate with an ultrasound aspirator, so it was carefully resected in several pieces. The overlying dura was also removed and sent for pathologic studies. The inner side of the bone flap was drilled before replacement.
Figure 2. Macroscopical view of the specimen.
A: One of the pieces resected was 8 x 6 x 2 cm in dimension.
It exhibited a smooth poly-lobulated surface mimicking cortical sulci and gyri.
B: When the specimen was cut, it showed a white-greyish firm homogenous tissue.
Within the first 72 hours after the intervention the patient suffered several generalised seizures that required additional anticonvulsant therapy (valproic acid plus phenytoin) for definite control. No neurological defect developed and the patient was discharged asymptomatic at the seventh day. On the basis of the histopathological specimens, a diagnosis of intracranial chondroma of the dural convexity was made. Microscopically, it exhibited well-differentiated cartilaginous cells within a fibrous matrix forming lobulated structures without any signs of malignancy (figure 3). Subsequent brain MRI studies have shown no evidence of recurrence after 33 months of follow up (figure 4). The patient is currently under two antiepileptic drugs (valproic acid and lamotrigine) but he suffers sporadic generalised seizures because of common incompliance with medication.

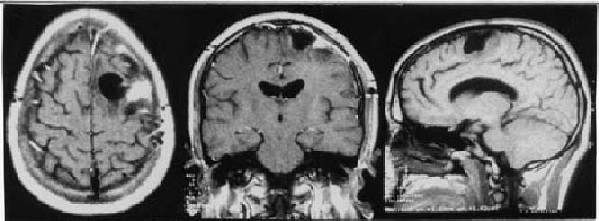
Figure 4. Postoperative MRI: axial, coronal and sagittal views after total excision
of the dural chondroma. Residual malacia is still present
after 33 months and may contribute to the persistence of epilepsy.
]]> Discussion
Chondromas are extremely rare intracranial tumours (less than 0.3% of all)3,11,16. They basically arise from the synchondrosis located at the skull base and they have been previously associated with Ollier's multiple enchondromatosis and Mafucci's syndrome8,19. They show equal sex distribution with a peak incidence in the third decade15,16. Only twenty-five cases of intracranial chondromas arising from the dural convexity are reported in the literature so far3,16.
Several hystopathogenetic theories have been proposed to explain the formation of such tumours. These are metaplasia of meningeal fibroblasts and perivascular meningeal tissue1,2,6, traumatic or inflammatory cartilaginous activation of fibroblasts16 and growth of aberrant embryonal cartilaginous rests in the dura mater (heterotopical condrocytes)6,20. It seems likely, although impossible to ascertain, that the existence of embryonic remnants of chondrogenic cells may explain the development of dural chondromas, as considered to be the origin of basal chondromas6,16.
Chondromas present clinical features somehow similar to meningiomas, even though they appear in a younger population13. Because of the slow growing nature of chondromas they become very large-sized at presentation13,16. The mass effect of these tumours may result in seizures or focal deficits, as well as, raised intracranial pressure or hydrocephalus3. Malignant degeneration has been reported before, especially in partially resected tumours12,15.
The radiological features of dural chondromas have been thoroughly reviewed by Nakayama et al16. Chondromas are known to be able to erode the inner side of the skull bone due to local tumour growth and they may induce hyperostosis3,15. CT scan imaging shows a mass of variable density appearance due to different degrees of calcification with minimum to moderate contrast enhancement's. The centre of the tumour may have low-density, reflecting necrosis or cystic degeneration. MRI studies show a wellcircumscribed lesion without surrounding tissue oedema, that exhibit heterogeneous signal with intermediate to low intensity on T1-weighted images and mixed intensity on T2-weighted images. A minimum enhancement after gadolinium administration is expected. Chondromas do not display any dural tail as in classic convexity meningiomas and the latter exhibit a more marked heterogeneous contrast enhancement. Angiogram is clue to differentiate from meningiomas since chondromas are completely avascular extracerebral space-occupying lesions2,11,15,16.
Complete tumour resection including its dural attachment should be the goal of surgery since no recurrence is expected to occur after total excision3,16,17. Long-term prognosis is favourable if no complications follow surgical removal1,9,15. Epilepsy may persist after tumour removal as in the case presented. Radiation therapy is currently not recommended for residual tumours or inoperable patients since chondromas do not clearly respond to irradiation and it may induce malignant degeneration3,7. As in classic meningiomas, surgical resection is easier when the tumour is located in the convexity compared to skull base chondromas3. Its clear subarachnoid plane allows fine dissection from the underlying brain. They are firm, almost completely avascular lesions not invasive or adherent to the surrounding tissues. These features favour their complete resection with a minimum morbidity. As in the case presented, very large tumours need piece by piece resection in order to avoid excessive brain retraction16.
Noonan's syndrome (also known as pseudo-Turner syndrome) is a complex familial genetic disorder with an autosomal dominant inheritance pattern, presenting in 1/1.000 people. It was first described in 1963 by a cardiologist (JA Noonan) and a paediatrician (D Ehmke) on the basis of several clinical and cytogenetic findings in a series of nineteen young patients of both sexes with various cardiac malformations, short stature, hypertelorism and skeletal anomalies18. Noonan's phenotype resembles that of Turner's syndrome but exhibits no chromosomal defect. Noonan's syndrome is caused by mutations at chromosome location 12q24.1, a gene encoding the non-receptor type 11 protein tyrosine phosphatase22. The case described presented with the typical phenotype including hypertelorism, short stature, retarded sexual development and mild mental retardation but showed no severe cardiac anomalies (non symptomatic pulmonary valve stenosis), even though his older brothers had died postpartum due to congenital heart malformations.
No predisposition of Noonan's syndrome for tumoural development is reported in the literature. In some patients, Neurofibromatosis type 1 (NF-1) demonstrates phenotypic overlap with Noonan's syndrome resulting in the so-called Neurofibromatosis-Noonan's syndrome, which represents a variant of NF-1, caused by mutations in the NF-1 gene5. Association of a dural convexity chondroma with Noonan's syndrome is unique as far as the literature is concerned. We can not offer a hystogenetic explanation for such association. Even though the concurrence of both may be incidental, we suggest that the presence of aberrant embryonal cartilage remnants could be a special feature accompanying the abnormal tissue development and/or cell migration in Noonan's syndrome, and the ultimate cause for the formation of a dural chondroma.
References
]]>1. Acampora, S., Troisi, F., Fusco, G., Del Gaizo, S.: Voluminous intracraneal condroma. Surg Neurol 1982; 18:254-257. [ Links ]
2.Ahyai, A., Spoeni, O.: Intracerebral chondroma. Surg Neurol 1979; 11: 431-433.
3. Colpan, E., Attar, A., Erekul, S., Arasil, E.: Convexity dural chondroma: a case report and review of the literature. J Clin Neurosci 2003; 10: 106-108.
4. De Coene, B., Gilliam, C., Grandin, C., Nisolle, J.F., Trigaux, J.P., Landou, J.B.: Inusual location of an intracranial condroma. AJNR Am J Neuroradiol 1997; 18: 573-575.
5. De Luca, A., Bottillo, I., Sarkozy, A., et al.: NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet 2005; 77: 1092-1101.
6. Dutton, J.: Intracranial solitary chondroma. Case report. J Neurosurg 1978; 49: 460-463.
7. Falconer, M.A., Bailey, I.C., Duchen, L.W.: Surgical treatment of chordoma and chondroma of the skull base. J Neurosurg 1968; 29: 261-275.
8. Ghogawala, Z., Moore, M., Strand, R., Kupsky, W.J., Scott, R.M.: Clival chondroma in child with Ollier's disease: case report. Pediatr Neurosurg 1991; 17: 53-56.
9. Hardy, R.W. Jr., Benjamin, S.P., Gardner, W.J.: Prolonged survival following excision of dural chondroma. J Neurosurg 1978; 48:125-127.
10. Ishibasi, H., Matsuno, H., Nagata, S., Onitsuka,H., Fukui, M.: Posterior fosse chondroma arising from the tentorium: a case report. Surg Neurol 1999; 52:604-606.
]]> 11. Kretzschmar, H.A., Eggert, H.R., Beck, U., Furmaier, R.: Intracranial chondroma. Case report. Surg Neurol 1989; 32: 121-125.12. Kurt, E., Beute, G.N., Sluzewski, M., van Rooij, W.J., Teepen, J.L.: Giant chondroma of the falx. Case report and review of the literature. J Neurosurg 1996; 85:11611164.
13. Lacerte, D., Gagne, F., Copty, M.: Intracranial chondroma. Report of two cases and review of the literature. Can J Neurol Sci 1996; 23: 132-137.
14. Luzardo-Small, G., Mendez-Martinez, O., Cardozo-Duran, J.: Cavitated (cystic) falcne condroma: case report. Br J Neurosurg 1999; 13: 426-428.
15. Mapstone, T.B., Wongmorengkolrit, T., Roessman, U., Ratcheson, R.A.: Intradural chondroma: a case report and review of the literature. Neurosurgery 1983; 12: 111-114.
16. Nakayama, M., Nagayama, T., Hirano, H., Oyoshi, T., Kuratsu, J.: Giant chondroma arising from the dura mater of the convexity. J Neurosurg 2001; 94: 331-334.
17. Nakazawa, T., Inoue, T., Suzuki F., Nakasu, S., Handa, J.: Solitary intracranial chondroma of the convexity dura: case report. Surg Neurol 1993; 40: 495-498.
18. Noonan, J.A.: Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am J Dis Child 1968; 116: 373-380.
19. Pospiech, J., Mehdom, H.M., Reinhardt, V., Grote, W.: Sellar chondroma in a case of Ollier's disease. Neurochirurgia (Stuttg)1989; 32: 30-35 (abstract).
20. Russell, D.S., Rubinstein, L.S.: Pathology of tumours of the nervous system. Baltimore, Williams & Wilkins, 1971; pp. 60-61.
]]> 21. Salazar, J., Vaquero, J., Aranda, I.F., Menéndez, J., Jiménez, M.D., Bravo, G.: Choroid plexus papilloma with chondroma: case report. Neurosurgery 1986; 18: 781-783.22. Tartaglia, M., Kalidas, K., Shaw, A., et al.: PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70:1555-1563.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. Pedro David Delgado López
Servicio de Neurocirugía. Hospital General Yagüe.
Avda. del Cid 96. 09005 Burgos.
Recibido: 16-05-06.
Aceptado: 6-06-06