
El laboratorio en el diagnóstico de las enfermedades raras
Laboratory diagnosis of rare diseases
R. Artuch, J. Moreno, R. M. Puig, M. Quintana, R. Montero, A. Ormazábal, M. A. Vilaseca
Servicio de Bioquímica. Departamento de errores congénitos del metabolismo. CIBERER. Hospital Sant Joan de Déu. Esplugues. Barcelona.
Dirección para correspondencia
]]>
RESUMEN
Los errores congénitos del metabolismo (ECM), constituyen un grupo de enfermedades cuyo diagnóstico se realiza habitualmente por medio de la cuantificación de metabolitos en fluidos biológicos. El objetivo de la presente revisión es describir el papel del laboratorio en el diagnóstico de los ECM haciendo especial énfasis en los procedimientos analíticos disponibles en la actualidad, revisando sus ventajas y también sus limitaciones. En conclusión, el número tan elevado de ECM que se está identificando hace necesaria la implementación progresiva de nuevas tecnologías en los laboratorios dedicados al estudio de estas enfermedades. Si bien es cierto que estas nuevas herramientas han dotado a los laboratorios de una mayor capacidad diagnóstica, no hay que olvidar pruebas convencionales que siguen siendo necesarias para este diagnóstico, así como toda una serie de variables que influyen en la recogida de las muestras y en la expresión de la enfermedad, que pueden conducir a fallos diagnósticos.
Palabras clave. Enfermedades raras. Errores congénitos del metabolismo. Análisis de metabolitos. Estudios genéticos. Pruebas funcionales.
ABSTRACT
Inborn errors of metabolism (IEM) are a group of diseases whose diagnosis is usually performed by quantification of several metabolites in biological fluids. The aim of this review is to report the role of the laboratory in IEM diagnosis, highlighting the methods available at present and their advantages and limitations. In conclusion, the huge number of recognized IEMs strongly advises the implementation of new high- output technologies in the laboratories devoted to IEM diagnosis. Although these technologies offer a high diagnostic ability, routine analyses are still very important, as well as consideration of several variables involved in biological sample collection and disease expression that may lead to misdiagnosis of IEMs.
Key words. Rare diseases. Inborn errors of metabolism. Metabolite analysis. Genetic studies. Challenge test.
Introducción
Las enfermedades raras son un conjunto muy heterogéneo de trastornos que obedecen a causas genéticas y no genéticas. Dentro de las enfermedades raras de base genética existe un subgrupo de alteraciones que son los errores congénitos del metabolismo (ECM), que constituyen un grupo de enfermedades cuyo diagnóstico se realiza habitualmente por medio de la cuantificación de metabolitos en fluidos biológicos en pacientes previamente seleccionados en base a la sospecha clínica1. Las alteraciones bioquímicas comprenden desde anomalías leves de la analítica básica que sugieren la existencia de un ECM y a veces permiten orientar el diagnóstico, hasta alteraciones muy importantes de los marcadores bioquímicos específicos (principalmente metabolitos) de cada una de estas enfermedades. El paso final en todo proceso diagnóstico de estas enfermedades es la identificación de las mutaciones causantes de la enfermedad en genes determinados2.
]]> Dada la heterogeneidad del conjunto de los ECM hemos considerado tres grandes grupos3,4, ya que esta clasificación tiene una cierta relación con el diagnóstico bioquímico de los ECM:1. Errores congénitos del metabolismo de moléculas complejas.
2. Errores congénitos del metabolismo intermediario o de las moléculas simples.
3. Errores congénitos del metabolismo energético.
En el primer grupo se incluyen ECM que implican las moléculas complejas localizadas en el lisosoma, peroxisoma, retículo endoplásmico o aparato de Golgi. Los síntomas clínicos que muestran acostumbran a orientar sobre el origen de dichos ECM3, que en algunos casos serán directamente confirmados enzimática y/o genéticamente. No obstante, existen algunos estudios específicos como el análisis de los ácidos grasos de cadena muy larga para el diagnóstico de enfermedades peroxisomales y el isoelectroenfoque de la transferrina para los defectos congénitos de la glicosilación, que permiten seleccionar los pacientes e incluso distinguir los diferentes tipos dentro de estas enfermedades4.
El segundo grupo incluye principalmente ECM intermediario, o de las moléculas simples, que dan lugar a un síndrome de intoxicación o a síntomas neurológicos puros, como aminoacidopatías, acidurias orgánicas, defectos del ciclo de la urea y del metabolismo de los carbohidratos5-7. En ellos, la alteración del metabolismo se traduce generalmente en anomalías de los perfiles bioquímicos en líquidos biológicos (sangre, orina y/o LCR), que orientan sobre las vías metabólicas afectadas y los puntos concretos de bloqueo, lo que permite confirmar los defectos por análisis enzimáticos o de caracterización de las proteínas mutadas y estudios genéticos.
El tercer grupo incluye los errores congénitos del metabolismo energético, que implican defectos de la cadena respiratoria mitocondrial, metabolismo del piruvato, glucogenosis hepáticas (I y III) y defectos de la β-oxidación de los ácidos grasos. Las alteraciones bioquímicas en algunos casos son específicas (β-oxidación) pero en muchos son inespecíficas del origen del ECM, aunque sí sugestivas del tipo de enfermedad8.
El objetivo de la presente revisión es describir el papel del laboratorio en el diagnóstico de los ECM, haciendo especial énfasis en los procedimientos analíticos disponibles en la actualidad, revisando sus ventajas y también sus limitaciones.
]]> Muestras biológicas para el diagnóstico de los ECM
Los especímenes utilizados para el estudio inicial de pacientes con sospecha de ECM son básicamente líquidos biológicos, como sangre, orina y, si es necesario, líquido cefalorraquídeo1. En la tabla 1 se detallan los análisis más habituales practicados en estas muestras para el diagnóstico de ECM.

El primer aspecto que debemos tener en cuenta para el diagnóstico de laboratorio de un ECM es qué tipo de muestra biológica vamos a estudiar, ya que deberíamos conocer hasta qué punto la enfermedad que buscamos se expresa en dicho material. No es raro que ciertos ECM no se puedan diagnosticar por medio del estudio bioquímico en sangre (por ejemplo, defectos del transporte tubular renal de aminoácidos, algunas acidurias orgánicas), o en orina (enfermedades mitocondriales, glucogenosis). En este apartado adquiere especial relevancia el líquido cefalorraquídeo, ya que su adecuada obtención es esencial para el diagnóstico de al menos 10 enfermedades que no se expresan en sangre y orina, como son defectos del transporte de glucosa al cerebro (Glut-1), deficiencias de la síntesis de dopamina y serotonina, defectos del metabolismo de las pterinas que cursan sin hiperfenilalaninemia, defectos del metabolismo de la vitamina B6, ciertas aminoacidopatías (hiperglicinemia no cetósica y defectos de serina), defectos del transporte de folato y algunos trastornos mitocondriales de expresión central (Tabla 1)9-11.
Un segundo aspecto muy importante para el diagnóstico de laboratorio de los ECM es que las muestras deben tomarse, a ser posible, en el momento de la descompensación metabólica (si la hay) y antes de iniciar cualquier tipo de tratamiento1, si bien esto no sería necesario en pacientes con ECM de moléculas complejas, por ejemplo. Para el laboratorio es muy útil que la petición de análisis se acompañe de información clínica sobre la edad, sexo, antecedentes familiares y personales de interés, breve resumen de la historia clínica, dietas especiales y medicación, ya que esto facilitará la interpretación del resultado12. Estos datos son indispensables ya que el diagnóstico definitivo no deriva en general de un dato bioquímico aislado, sino de la interpretación del conjunto de datos bioquímicos y clínicos del paciente, en especial en el contexto del momento de la recogida de la muestra cuando se trata de errores del metabolismo que cursan con descompensaciones. Por ello, la colaboración clínico-bioquímico es indispensable para mejorar la eficacia del diagnóstico de estas enfermedades13.
El tercer aspecto que es importante tener en cuenta respecto a las muestras es el tipo de análisis que es necesario solicitar. Es útil disponer de perfiles de pruebas bioquímicas a solicitar en sangre, orina y LCR, que incluyan las determinaciones que consideremos útiles en el diagnóstico de un ECM, para que los especímenes (que a veces son muestra única con muy poco volumen) se recojan de la forma adecuada y sean aprovechados al máximo, ya que se trata con frecuencia de neonatos o pacientes en una situación clínica grave14-15. Es importante además incluir analíticas rutinarias básicas, ya que muchas veces son las que pueden orientar mejor la enfermedad. En la tabla 1 se clasifican los análisis en básicos (o rutinarios) y específicos asociando la información que pueden aportar respecto al diagnóstico de los ECM. Los tejidos y muestras de ADN necesarios para la confirmación diagnóstica pueden recogerse en una segunda etapa, cuando la orientación del origen del ECM se haya alcanzado mediante el análisis de líquidos biológicos2.
]]> Procedimientos bioquímicos para el diagnóstico de los ECM
Entre los análisis bioquímicos (y de laboratorio en general) aplicados para el diagnóstico de un ECM, distinguimos unos procedimientos simples que constituyen el análisis básico de alteraciones metabólicas, que aporta una primera orientación sobre la vía afectada. Estos procedimientos están disponibles en todos los laboratorios de análisis clínicos, y la mayoría de ellos están automatizados, por lo que obtendremos un respuesta rápida y fiable del resultado.
El segundo paso consiste en realizar otros análisis específicos más indicativos de la enfermedad en concreto, pero por ello también más sofisticados o sin posibilidad de momento de automatización y realización rutinaria1. Idealmente, estos estudios deberían ser practicados en las mismas muestras de sangre y orina recogidas en descompensación (y en las que se practicaron ya las determinaciones más rutinarias) y dependiendo de la sospecha clínica y de los datos aportados en los análisis básicos, elegir los más adecuados.
Se revisan los diferentes procedimientos de análisis disponibles en laboratorios que se dedican al diagnóstico de los ECM, sin especificar sus aspectos técnicos.
Análisis de metabolitos
Métodos espectrofotométricos
Esta metodología está disponible desde hace varias décadas en todo tipo de laboratorios, y en los últimos años se han ido automatizando para el análisis de los parámetros más rutinarios. Se basan en medir la concentración de un sustrato (o la actividad de una enzima) aplicando la respuesta diferente que tienen las moléculas ante una longitud de onda de luz determinada. Este procedimiento sigue siendo muy útil para la determinación de moléculas concretas (por ejemplo, carnitina, tiosulfato, etc), pero especialmente para la determinación de actividades enzimáticas en plasma o tejidos (por ejemplo, determinación de la actividad biotinidasa en suero o de enzimas de la cadena respiratoria mitocondrial en biopsia de músculo). Habitualmente, los procedimientos espectrofotométricos no son útiles cuando hay que determinar varios metabolitos con unas características químicas similares (por ejemplo, aminoácidos o ácidos orgánicos), y hay que recurrir a métodos de separación e identificación más especiales.
Métodos de separación e identificación de metabolitos
Cromatografía líquida
Esta metodología también ha sido aplicada en diferentes laboratorios desde hace décadas, y especialmente en los que se han dedicado al diagnóstico de los ECM16. Se basa en separar moléculas según sus diferentes propiedades físico-químicas, utilizando un solvente (líquido o fase móvil) y un soporte (habitualmente una columna de cromatografía). Las moléculas se separan gracias a las interacciones que ejercen con la fase móvil y la columna de cromatografía, y después se detectan y cuantifican utilizando diferentes sistemas (ver más adelante). Existen diferentes tipos de cromatografía líquida. Únicamente citar, por su grandísima versatilidad, la cromatografía líquida de alta presión (HPLC), que está basada en aplicar una elevada presión al sistema para conseguir unos análisis rápidos y reproducibles. Una vez que se han separado los diferentes metabolitos, existe la posibilidad de detectarlos y cuantificarlos por diferentes detectores como por ejemplo, espectrofotométricos16, de fluorescencia17 (aprovechando la fluorescencia natural de algunas moléculas, o de reactivos fluorescentes que se ligan a dichas moléculas), electroquímicos18 (aplicando potenciales de oxidación y reducción) o de espectrometría de masas17. Este último sistema ha cambiado sustancialmente la actividad diagnóstica en el campo de los ECM. La cromatografía líquida sigue siendo muy útil para la determinación de múltiples metabolitos, como por ejemplo aminoácidos, vitaminas, neurotransmisores y otros tipos de moléculas.
]]> En la figura 1 se presentan diferentes cromatogramas obtenidos por cromatografía líquida en controles sanos y pacientes con diferentes ECM. Dentro del laboratorio de diagnóstico de los ECM, el análisis de aminoácidos por cromatografía líquida tiene una gran importancia, ya que con una sola determinación cromatográfica se pueden diagnosticar muchas enfermedades1. La única limitación de estos procedimientos es que suelen ser costosos de realizar en tiempo, la preparación de la muestra antes del análisis requiere procedimientos laboriosos, son difícilmente automatizables y, por tanto, no son idóneos para respuestas inmediatas en un elevado número de muestras.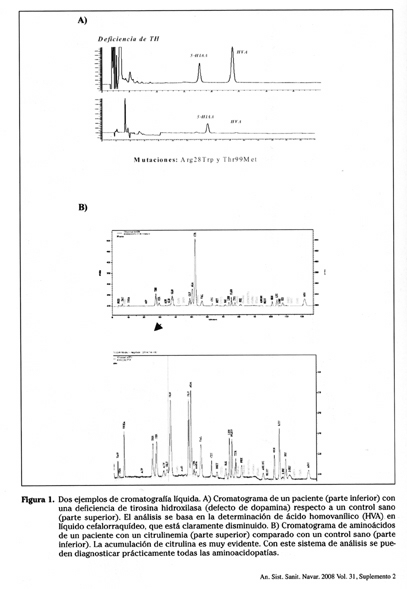
Cromatografía de gases
Esta metodología también ha sido aplicada en laboratorios dedicados al diagnóstico de los ECM desde hace décadas19. Se basa en separar moléculas volátiles, utilizando una fase móvil gaseosa (el gas transporta los metabolitos volátiles) y un soporte (habitualmente son columnas capilares de 30 metros de longitud). Las moléculas se separan y se detectan y cuantifican utilizando diferentes sistemas, entre los que destaca, por su enorme utilidad en el diagnóstico de los ECM, la espectrometría de masas19. Este procedimiento se basa en identificar la molécula, que previamente hemos separado por cromatografía de gases, gracias a diferentes iones en los que se descompone cada molécula de forma única, con lo cual se identifican de manera inequívoca. Este procedimiento es especialmente útil para la determinación de los ácidos orgánicos en orina, pero también de otros componentes. Con este procedimiento se pueden diagnosticar unos 130 ECM distintos, aunque esto requiere la estandarización de diferentes métodos que habitualmente son complejos de realizar e interpretar19. A pesar de que este procedimiento tiene una capacidad diagnóstica elevada en manos expertas, puede arrojar resultados falsos negativos, especialmente si la muestra no se ha recogido en el momento de la descompensación, por ejemplo. Por ello es importante dar el mensaje de que éste y otros procedimientos más sofisticados pueden estar sujetos a las variaciones en las concentraciones de los metabolitos debido a la evolución episódica de una enfermedad concreta.
Espectrometría de masas en tándem acoplada a cromatografía líquida (LC-MS/MS)
Este procedimiento de análisis ha experimentado un extraordinario avance en la última década, y está plenamente relacionado con el concepto de metabolómica20. A diferencia de los procedimientos anteriormente descritos, la preparación de la muestra antes del análisis y la detección y cuantificación de los diferentes metabolitos se realizan de forma más rápida17,20 . El sistema consiste en un cromatógrafo líquido que introduce las muestras en el detector de masas (cuadrupolo en tándem). Por tanto, es idóneo para análisis de múltiples metabolitos no volátiles de forma simultánea en muestras biológicas (principalmente sangre seca y orina). La posibilidad de determinar todos estos metabolitos en sangre seca hace que este método sea idóneo para los programas de detección precoz de ECM. De hecho, en varios países de Europa y en algunas comunidades de España ya se está aplicando para el diagnóstico precoz de más de 30 ECM, en contraposición con los sistemas actuales de detección de la fenilcetonuria, hipotiroidismo y fibrosis quística21. Además, permite el diagnóstico dirigido de otras muchas enfermedades metabólicas, por lo que hoy en día constituye una herramienta esencial para el diagnóstico de los ECM20. No obstante, todavía es un procedimiento complejo (en especial la interpretación de los resultados), y costoso, por lo que de momento sólo se está implantando en centros de referencia para diagnóstico de los ECM, pero no en los laboratorios clínicos de la mayoría de los hospitales. Por otro lado, conviene recordar que no son procedimientos infalibles, ya que en el caso de algunos ECM del metabolismo intermediario que se manifiestan principalmente durante la descompensación del paciente, el análisis de una muestra no adecuada puede llevar también a resultados falsos negativos. En la tabla 2 figuran algunas de las enfermedades que se pueden diagnosticar por medio de LC-MS/MS.

Si la combinación de los datos clínicos y bioquímicos obtenidos mediante los métodos de laboratorio básicos y los específicos anteriormente citados ha permitido una orientación definitiva del defecto, se procede a la realización de estudios de confirmación de la proteína afectada por la mutación del ADN que constituye el origen de la enfermedad. Estos estudios de confirmación enzimática o de caracterización de la proteína alterada, pueden realizarse en suero, leucocitos, eritrocitos, fibroblastos cultivados procedentes de biopsia de piel o en tejidos (músculo, hígado, riñón, corazón, etc). Los estudios genéticos de las mutaciones implicadas en el defecto completaran el estudio.
Análisis de proteínas
]]> El paso final en todo proceso diagnóstico de los ECM es la identificación de las mutaciones causantes de la enfermedad en genes determinados2. Estos genes codifican proteínas con diversas funciones (enzimas, transportadores, etc.) por lo que el análisis de proteínas muchas veces también es esencial en el proceso diagnóstico especialmente cuando tras el análisis de metabolitos no se puede discernir qué gen está implicado en la enfermedad (por ejemplo, la determinación del perfil de sialotransferrina como paso inicial en los defectos congénitos de la glicosilación). Otras veces, se desconoce el marcador molecular de la enfermedad, y sólo es posible identificar un fallo en la expresión o función de la proteína como diagnóstico del ECM. Esto ocurre habitualmente en grupos de enfermedades complejas, como en el caso de las encefalomiopatías mitocondriales22.El análisis de proteínas ha experimentado un gran avance en la última década, especialmente gracias a los estudios de proteómica que permiten analizar la expresión de muchas proteínas de forma simultánea20. No obstante, estos sistemas de análisis no suelen estar disponibles en el laboratorio clínico, por lo que revisaremos aquellos procedimientos que permiten el estudio de proteínas específicas implicadas en los ECM. Hay múltiples métodos automatizados de cuantificación de proteínas (nefelometría, quimioinmunoluminiscencia) disponibles en los laboratorios automatizados que no serán objeto de este trabajo.
Cuando se va a realizar un análisis de proteínas, tenemos que discernir entre dos posibilidades fundamentalmente. Estudiar la función de la proteína y la expresión de la misma. Los procedimientos aplicados son diferentes, y la información que aportan también. Conviene remarcar que la mayor parte de las veces necesitaremos células para dicha determinación, por lo que habrá que plantear la realización de una biopsia cuando proceda.
Estudios de función proteica
Se basan en medir la función de determinada proteína. Si esta proteína es un enzima, se medirá la actividad enzimática habitualmente por espectrofotometría, incubando la proteína con los sustratos adecuados22. También es posible medir la actividad de una o varias proteínas de una vía metabólica completa incubando las células con sustratos marcados radiactivamente, y medir la formación de productos específicos de esa vía metabólica por procedimientos radiométricos22. Este tipo de estudios de oxidación de sustratos marcados son muy útiles para determinar la función de una vía metabólica completa (por ejemplo oxidación de piruvato para evaluar la función energética mitocondrial) y muchas veces aportan datos no observados anteriormente sobre el diagnóstico de un ECM. Si se pretende estudiar la función de un transportador (por ejemplo Glut-1 o transportador de creatina), se analizará en las células en las que se exprese esta proteína la incorporación del sustrato (glucosa y creatina marcadas) al interior de las mismas.
Estudios de expresión proteica
En ocasiones, es más útil evaluar la expresión de la proteína (cantidad, peso molecular, isoformas) que la propia función23. No obstante, conviene recordar en este punto que muchas de las mutaciones causantes de ECM no producen un descenso en la cantidad total de la proteína, ni alteran su peso molecular, por lo que la utilidad de estas pruebas es más limitada en estos casos. Estas pruebas se recomiendan cuando las mutaciones causantes son grandes deleciones que reducen el tamaño de la proteína o, cuando la mutación causa inestabilidad de la proteína y se reduce la cantidad total. También son muy útiles, como en el caso de los defectos congénitos de la glicosilación, cuando una mutación en uno de los cientos de genes que regulan este complejo proceso altera la glicosilación de una proteína en concreto (sialotransferrina)23,24. Las pruebas más habituales para el estudio de la expresión de la proteína son los sistemas de separación de proteínas. Existen varias metodologías disponibles para este fin (electroforesis, cromatografía, etc.). Algunas de ellas como la electroforesis son métodos tradicionales, desarrollados ya hace varias décadas, pero que siguen siendo muy útiles para el diagnóstico de los ECM. Se basan en general en estudiar la proteína sometida a un campo eléctrico, que hará migrar diferencialmente a las proteínas según la carga neta de la misma (ioelectroenfoque, electroforesis capilar) o de acuerdo a su peso molecular (Western-blot)23. En la figura 2 se detalla un análisis de sialotransferrina por Western-blot y por isoelectroenfoque de un paciente con defecto congénitos de la glicosilación.
Por último, se han implantado en los últimos años sistemas de análisis de proteínas de alto rendimiento (Maldi-TOF). Existen excelentes revisiones de esta materia en la bibliografía24.
Estudios genéticos
]]> Hace unos años sólo unos pocos centros tenían la tecnología necesaria para afrontar este tipo de estudios. Con los avances tecnológicos, muchos de estos métodos están ya disponibles en el laboratorio clínico de diagnóstico de ECM. Hay que destacar que el diagnóstico genético es el paso final en el estudio de los ECM2,8. Cada vez es más frecuente que no sea necesario estudiar la función o expresión de una proteína, y se puede pasar al diagnóstico genético directo incluso en ADN obtenido de sangre periférica2. Esto ocurre cuando el perfil de metabolitos es suficientemente claro, o bien cuando no es posible biopsiar el tejido donde se expresa la proteína (por ejemplo, en las deficiencias de la síntesis de dopamina causadas por defectos genéticos de la enzima tirosina hidroxilasa, que sólo se expresa en neuronas dopaminérgicas y en la médula adrenal)10. En la tabla 3 se citan algunas de las pruebas genéticas más frecuentemente utilizadas con una breve explicación de sus fundamentos.
Merece una descripción un poco más detallada la secuenciación. Consiste en la separación de fragmentos del gen que se va a estudiar (previamente amplificado por PCR) por electroforesis capilar y detección de fluorescencia inducida por láser. Existen sistemas automatizados que permiten el estudio de un gran número de mutaciones en pacientes con sospecha de ECM2, y estos estudios se pueden realizar en ADN obtenido de sangre periférica. A pesar de estos avances, hay que remarcar que esta prueba también tiene sus limitaciones, y un resultado negativo nunca se debe considerar como definitivo si hay una sospecha clínica y bioquímica razonable. Es decir, a pesar de ser probablemente la herramienta diagnóstica de mutaciones más potente, no identifica determinadas alteraciones (como deleciones por ejemplo), por lo que habrá que recurrir a procedimientos alternativos.
Pruebas funcionales
No es raro que en el proceso diagnóstico de los ECM no se disponga de la información clínica y bioquímica suficiente como para sospechar la causa genética de la enfermedad8,25. Esto ocurre esencialmente en aquellos ECM que cursan de forma episódica, con brotes de descompensación metabólica (hipoglucemias por defectos de la beta-oxidación o por glucogenosis, hiperamoniemias secundarias a defectos leves del ciclo de la urea, etc). El fundamento de las pruebas funcionales consiste en causar una descompensación metabólica leve en el paciente, que debe estar estrictamente monitorizada y obtener las muestras (sangre y orina) para análisis bioquímicos siguiendo unos protocolos8,25. De esta forma, podemos revelar la acumulación de ciertos metabolitos claves para el diagnóstico que en condiciones basales no ha podido ser demostrada. Las pruebas funcionales más frecuentemente aplicadas son:
Prueba del alopurinol y de la sobrecarga proteica. Indicada para diagnóstico de defectos del ciclo de la urea.
Sobrecarga de glucosa, para el diagnóstico de encefalomiopatías mitocondriales.
Prueba del ejercicio y de la isquemia para el diagnóstico de encefalomiopatías mitocondriales y de glucogenosis musculares
]]> Prueba del ayuno, para diagnóstico diferencial de las hipoglucemias. Sobrecarga de galactosa y fructosa, para el diagnóstico de galactosemias y fructosemias.
Algunas de estas pruebas no están exentas de cierto riesgo para el paciente, por lo que sólo se deberán realizar cuando sea necesario, de forma protocolizada y bajo un estricto control médico durante toda la duración de la prueba8,25.
Protocolo de exitus en pacientes con sospecha de ECM
Conviene también disponer de un protocolo de exitus ante la sospecha de ECM, que permita alcanzar el diagnóstico post-mortem y dar el adecuado consejo genético a las familias (Tabla 4)25. Es útil acompañarlo de un kit de exitus que incluya el protocolo, los tubos adecuados, el papel de filtro para impregnar con sangre, un punch (aguja para obtención de biopsia de piel) y el medio de cultivo para la biopsia de piel. Esta muestra es especialmente importante para muchos diagnósticos de ECM, ya que permite cultivar y almacenar fibroblastos para estudios posteriores. Se puede conservar en un refrigerador en las unidades de Neonatos, UCI y Urgencias para facilitar la recogida correcta de las muestras en caso de exitus.

Conclusiones
Durante la última década se ha producido un avance espectacular en los procedimientos diagnósticos de los ECM. El número tan elevado de ECM que se está identificando hace necesaria la implementación progresiva de nuevas tecnologías de alto rendimiento diagnóstico en los laboratorios dedicados al estudio de estas enfermedades. Si bien es cierto que estas nuevas herramientas han dotado a los laboratorios de una mayor capacidad diagnóstica, no hay que olvidar pruebas convencionales que siguen siendo necesarias para este diagnóstico, así como controlar toda una serie de variables que influyen en la recogida de las muestras y en la expresión de la enfermedad que pueden conducir a fallos diagnósticos.
]]>Bibliografía
1. Sanjurjo P, Aquino L, Aldámiz-Echeverría L. Enfermedades congénitas del metabolismo: generalidades, grupos clínicos y algoritmos diagnósticos. En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA 2001: 29-52. [ Links ]
2. Desviat LR, pérez B, Ugrate M. Bases moleculares de las enfermedades metabólicas hereditarias. En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA 2001: 1-13. [ Links ]
3. Rebage V, López-Pisón J, Baldellou A. Errores congénitos del metabolismo en el período neonatal. En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA 2001: 53-66. [ Links ]
4. Saudubray JM, Charpentier C. Clinical phenotypes:diagnosis /algorithms. En Scriver CR,B udet Al, Sly WS, Valle D editors. The metabolic bases of inherited Disease. New-York: Mc Graw-Hill 2001: 1327-1403. [ Links ]
5. Baldellou A, López-Pisón J, Rebage V, Salazar MI. Errores congénitos del metabolismo de presentación precoz. An Esp Pediatr 1998; 114: 20-24. [ Links ]
6. Campistol J. Enfermedades metabólicas de presentación neonatal. Arch Pediatr 1995; 46: 115-117. [ Links ]
7. García-Silva MT. Errores congénitos del metabolismo con repercusión sobre el sistema nervioso en el recién nacido. Cuando y cómo investigarlos. Rev Neurol 2000; 31: 604-616. [ Links ]
8. Sanjurjo P, Aldámiz-Echeverría L, Aquino L. Pruebas funcionales en el estudio de los errores innatos del metabolismo. En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA, 2001: 73-79. [ Links ]
9. Ormazabal A, Oppenheim M, Serrano M, García-Cazorla A, Campistol J, Ribes A et al. Pyridoxal 5-phosphate values in cerebrospinal fluid: Reference values and diagnosis of PNPO deficiency in paediatric patients. Mol Genet Metab. 2008; 20 (en prensa). [ Links ]
10. Ormazabal A, García-Cazorla A, Fernández Y, Fernández-Alvarez E, Campistol J, Artuch R. HPLC with electrochemical and fluorescence detection procedures for the diagnosis of inborn errors of biogenic amines and pterins. J Neurosci Methods 2005; 142: 153-158. [ Links ]
11. Ormazabal A, García Cazorla A, Pérez Dueñas B, Pineda M, Ruiz A, López Laso E et al. Usefulness of analysis of cerebrospinal fluid for the diagnosis of neurotransmitters and pterin defects and glucose and folate transport deficiencies across blood brain barrier. Med Clin (Barc). 2006; 127: 81-85. [ Links ]
12. Pineda M, Vilaseca MA, Cardo E. Management of paediatric metabolic emergencies in intensive care. Care Crit Ill 1998; 14: 271-274. [ Links ]
13. Roth KS. Inborn errors of metabolism: the essentials of clinical diagnosis. Clin Pediatr 1991; 30: 183-190. [ Links ]
14. Leonard JV, Morris AAM. Inborn errors of metabolism around time of birth. Lancet 2000; 356: 583-587. [ Links ]
15. Ogier H, Saudubray J-M. Maladies héréditaires du metabolism à révélation aigüe néonatale: prise en charge, diagnostic et thérapeutique. En: Saudubray JM, editor. Maladies métaboliques. Progrès en pédiatrie. Paris: Doin éditeurs 1991: 63-81. [ Links ]
16. Tavazzi B, Lazzarino G, Leone P, Amorini AM, Bellia F, Janson CG et al. Simultaneous high performance liquid chromatographic separation of purines, pyrimidines, N-acetylated amino acids, and dicarboxylic acids for the chemical diagnosis of inborn errors of metabolism. Clin Biochem 2005; 38: 997-1008. [ Links ]
17. Arias A, Ormazabal A, Moreno J, González B, Vilaseca MA, García-Villoria J et al. Methods for the diagnosis of creatine deficiency syndromes: a comparative study. J Neurosci Methods 2006; 156: 305-309. [ Links ]
18. Montero R, Sánchez-Alcázar JA, Briones P, Hernández AR, Cordero MD, Trevisson E et al. Analysis of Coenzyme Q(10) in muscle and fibroblasts for the diagnosis of CoQ(10) deficiency syndromes. Clin Biochem 2008 (en prensa). [ Links ]
19. Kuhara T. Gas chromatography-mass spectrometric urinary metabolome analysis to study mutations of inborn errors of metabolism. Mass Spectrometry Rev 2005; 24: 814-827. [ Links ]
20. Röschinger W, Olgemöller B, Fingerhut R, Liebl B, Roscher AA. Advances in analytical mass spectrometry to improve screening for inherited metabolic diseases. Eur J Pediatr 2003; 162: S67-S76. [ Links ]
21. Pampols T, Maya A. Diagnóstico prenatal y detacción sistemática neonatal de las enfermedades congénitas del metabolismo. En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA 2001: 15-28. [ Links ]
22. Arenas J, Martin MA, Campos Y. Déficits bioquímicos de la cadena respiratoria mitocondrial. En: En: Sanjurjo P, Baldellou A editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon SA 2001: 405-410. [ Links ]
23. Artuch R, Ferrer I, Pineda J, Moreno J, Busquets C, Briones P et al. Western blotting with diaminobenzidine detection for the diagnosis of congenital disorders of glycosylation. J Neurosci Methods 2003; 125: 167-171. [ Links ]
24. Lacey JM, Magera MJ, Matern D, OBrien JF. A method for the determination of transferrin isoforms by liquid chromatography-mass spectrometry. J Inherit Metab Dis 2000; 23: 179. [ Links ]
25. Fernandes J, Saudubray JM. Diagnostic procedures: Function test and postmostem protocol. En: Fernandes J, Saudubray JM, van der Berge G, editors. Inborn metabolic diseases. Berlin: Springer-Verlag 1996: 41-46. [ Links ]
![]() Dirección para correspondencia: ]]>
Rafael Artuch Iriberri
Dirección para correspondencia: ]]>
Rafael Artuch Iriberri
Hospital Sant Joan de Déu
Departamento de Bioquímica Clínica
Passeig Sant Joan de Déu, 2
08950 Esplugues (Barcelona)
Tfno. +34932806169
Fax. +34932803626
E-mail: rartuch@hsjdbcn.org