Acidemia Metilmalónica
Methylmalonic Acidemia
Eva Buller Viqueiraa, Francisca Muñoz Peraltab y Juana Cabello Pulidoc
a Centro de la salud Virgen de la Oliva. Vejer de la Frontera, Cádiz (España).
b Centro de Salud Gonzalo Pérez Fabra. Paterna de Rivera. Cádiz (España).
c Centro de salud Rodríguez Arias. San Fernando. Cádiz (España).
RESUMEN
Presentamos el caso de una mujer de 20 años que acude a nuestra consulta por tics motores y fónicos que empeoran con el nerviosismo y desaparecen con el sueño y maniobras de distracción. Se deriva al hospital y se decide su ingreso. Al revisar sus antecedentes observamos que había sido ingresado en varias ocasiones durante su infancia antes de ser diagnosticada a los cuatro años de edad de acidemia metilmalónica. La sintomatología de esa patología comenzó durante su primera semana de vida, manifestando vómitos, hipotonía y fallo de medro. La acidemia metilmalónica es una enfermedad congénita que provoca un defecto a nivel de la enzima metilmalonil CoA-mutasa, ocasionando como consecuencia un acúmulo del ácido metilmalónico. Se diagnostica por aumento de ácido metilmalónico en orina y se confirma mediante análisis molecular genético. Durante este último ingreso la paciente fue diagnosticada de un tourettismo, sin determinar si fue secundario al tratamiento o si se trata de una manifestación crónica de su patología. La acidemia metilmalónica es una enfermedad rara de difícil diagnóstico tanto por su clínica inespecífica como por el escaso conocimiento sobre la misma por parte del personal sanitario.
Palabras clave: Metilmalonil-CoA mutasa. Ácido metilmalónico. Errores innatos del metabolismo. Aminoácidos.
ABSTRACT
The following is the case of a 20-year-old woman in seek of professional help because of chronic motor and phonic tics which worsened when nervous and disappeared when sleeping or when distraction manoeuvres were employed. She was referred to a hospital where she was hospitalized. In examining her medical history, we found that she required hospitalization on a number of occasions during childhood before being diagnosed with methylmalonic acidemia at the age of four. Her symptoms began within the first week of life, suffering from vomiting, hypotonia, failure to thrive and asthenia. Methylmalonic acidemia is a congenital disease which provokes a defect in the enzyme methylmalonyl-CoA mutase therefore causing a build-up of methylmalonic acid. It is diagnosed by an increase of methylmalonic acid in urine samples and it is confirmed with a molecular genetic analysis. In addition to this rare illness, the patient was also diagnosed with tourettism, without determining if it was due to treatment or a chronic presentation of her pathology. Methylmalonic acidemia is a rare disease, and consequently difficult to diagnose because of its unspecific clinical manifestations and because of healthcare workers' limited awareness of it.
Key words: Methylmalonyl-CoA mutase. Methylmalonic acid. Metabolism, inborn errors. Amino acids.
]]> Introducción
La acidemia metilmalónica (AMM) es una enfermedad metabólica innata, caracterizada por una escasa o nula actividad de la metilmalonil CoA mutasa (MCM). Al cabo de poco tiempo tras el nacimiento aparece deterioro generalizado, acidosis metabólica e hiperamonemia1. Puede provocar deterioro neurológico, insuficiencia renal o incluso la muerte, salvo los sujetos que responden a la vitamina B12. El tratamiento se basa fundamentalmente en disminuir la ingesta de proteínas de la dieta. En otras ocasiones, la clínica comienza a cualquier edad con un cuadro clínico más heterogéneo.
Actualmente conocemos que la AMM es una enfermedad rara de herencia autosómica recesiva, con una incidencia de entre 1 y 9 por 100.000 nacidos vivos2. Existen zonas con mayor incidencia como Arabia Saudí, donde afecta a 1 entre 2000 y 5000 nacidos vivos. Sin embargo, esta tasa podría ser mayor, pues podrían existir casos de muerte antes de la sospecha de la patología. Existen grupos étnicos que presentan mutaciones específicas con mayor frecuencia como es la japonesa o caucásicos descendientes de franceses y turcos3.
El retraso del diagnóstico de esta patología no es infrecuente tanto por su clínica inespecífica como por la dificultad para reconocerla por parte de los profesionales al tratarse de una enfermedad rara.
Observaciones clínicas
Presentamos el caso de una mujer de 20 años con diagnóstico de acidemia metilmalónica. Un mes tras el nacimiento de su segundo hijo comienza con movimientos involuntarios e irregulares en miembros superiores, cabeza y tronco además de tics fónicos emitiendo sonidos deglutorios, motivo por el que acude a consulta. Empeora con el nerviosismo y desaparecen con el sueño y maniobras de distracción. En este momento no existe una descompensación analítica. Se deriva al hospital y allí se decide su ingreso con el diagnóstico de trastorno del movimiento, posible corea secundaria a descompensación metabólica por su enfermedad de base (AMM) versus origen psicógeno.
Respecto a la AMM, la paciente presentó sus primeros síntomas relacionados con esta enfermedad entre la semana y el mes de vida. Comenzó con vómitos, hipotonía, fallo de medro y astenia, precisando varios ingresos hospitalarios y siendo diagnosticada en todas las ocasiones de gastroenteritis. Posteriormente, es el equipo de atención primaria el que realiza el seguimiento, siguiendo los controles que le corresponden por edad donde se vigila somatometría y objetivos psicomotrices. No se observan alteraciones, aunque en varias ocasiones refiere la madre que presenta menos actividad que otros niños de la misma edad y, aunque alcanza los objetivos psicomotrices para su edad a tiempo normal, no los practica por cansancio. Finalmente, a los 4 años de edad es diagnosticada de AMM, tras la realización de pruebas más específicas como la detección de aumento de ácido metilmalónico en orina y análisis molecular genético en el hospital de referencia, permaneciendo ingresada para su estudio. Al alta comienza con dieta pobre en proteínas con suplementos, mejorando significativamente su calidad de vida y sin presentar complicaciones asociadas. Es controlada en consulta de atención primaria desde entonces.
Hace tres años que abandonó el hogar familiar, la dieta y el tratamiento. Ha requerido desde entonces un ingreso hospitalario en unidad de cuidados intensivos por sepsis urinaria grave secundaria a pielonefritis aguda por Escherichia coli y varios ingresos por vómitos y fracaso prerrenal agudo. Diagnosticada también de hipertensión arterial esencial y ligera insuficiencia renal crónica.
Vuelve a acudir a su médico de familia por su primera gestación que cursa con hiperemesis y fue tratada con metoclopramida. Su hija era sana. Es seguida también por el equipo de atención primaria durante la segunda gestación que cursó también con hiperemesis y precisó metoclopramida. En esta ocasión su enfermedad de base se descompensó (bicarbonato elevado, homocisteína y ácido metilmalónico en orina elevado, carnitina libre y C3 carnitina elevados, resto de valores normales). Un mes más tarde, tras el nacimiento del bebé, comienza con movimientos involuntarios e irregulares como ya se ha descrito, pero en este momento no existe una descompensación analítica (gasometría venosa, amonio, sistemático de orina, función renal, iones, transaminasas y hemograma todos dentro de los límites de la normalidad). Se deriva al hospital y es ingresada con el diagnóstico de trastorno del movimiento, posible corea secundaria a descompensación metabólica versus origen psicógeno.
]]> Actualmente, presenta tics motores y fónicos que interfieren con su actividad diaria para vestirse, para el aseo y para comer. El nivel de tic es elevado con paso al acting out de tipo heteroagresivo verbal. Finalmente diagnosticada de tourettismo. A día de hoy no se ha establecido si es secundario al tratamiento prolongado con metoclopramida o si es un efecto secundario tardío de la AMM.En cuanto a la AMM, actualmente persisten las trasgresiones dietéticas, pero en menor medida, y acude ocasionalmente a consulta de enfermería y del médico de familia para seguimiento de su patología. Sigue tratamiento con clonazepam 500 mcg, carnitina 1 g, folinato calcio 15 mg, calcifediol 266 mcg y enalapril 20 mg.
Discusión
En 1987 Oberholzer, Stokke y colaboradores describieron un grupo de niños con grandes cantidades de ácido metilmalónico en sangre, orina y líquido cefaloraquídeo4. El metilmalonil coenzima A debe pasar a succinil coenzima A con ayuda de la MCM y su cofactor la cobalamina. Puede existir una alteración genética que provoque alteraciones en la síntesis de adenosilcobalamina, beneficiándose estos casos del tratamiento con vitamina B12. También existe el déficit parcial y completo de la MCM, siendo estos casos resistentes a la vitamina B12 y su gen se localiza en el cromosoma 6 p12-p21.2. Como consecuencia existe un defecto en el catabolismo de la metionina, treonina, valina e isoleucina, ocasionando un defecto de su metabolización y, por tanto, se acumula el ácido metilmalónico y, secundariamente, el amonio y lactato.
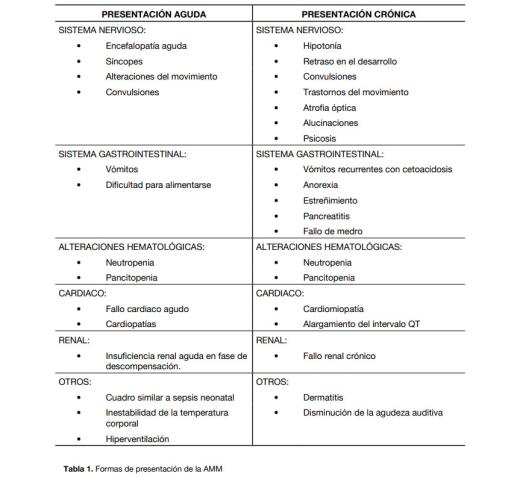
La clínica se presenta generalmente de dos formas5 (tabla 1), pero todos cursan con un periodo libre de enfermedad y muestran episodios de descompensación metabólica, que suelen asociarse a cuadros de infecciones o estrés. En la forma neonatal existe un periodo libre de síntomas que posteriormente comienza con un curso agudo y severo. La forma tardía comienza a partir del año de vida y puede ser, a su vez, aguda recurrente, intermitente (se exacerba por cuadros infecciosos, inmunización, cirugía, ayuno, trasgresión dietética, estrés), crónica progresiva o lentamente progresiva (los síntomas son desde insidiosos a asintomáticos). Queremos destacar los trastornos del movimiento que en fase tardía presentan una frecuencia de aparición de entre el 30-45 %5.
Ante la sospecha clínica es fundamental plantearnos este diagnóstico. Existen datos analíticos que nos pueden hacer sospechar su presencia como son la acidosis metabólica, la cetonemia o cetonuria, una hiperamonemia y el lactato normal o elevado. También disponemos de pruebas analíticas más específicas como el aumento de la C3 carnitina en sangre, del metilmalonil carnitina en sangre y de la homocisteína en sangre (también puede mostrar valores normales). El diagnóstico diferencial se realiza mediante el análisis de orina, donde podemos encontrarnos con niveles elevados de metilmalónico, metilcitrato, propionato o propionilglicina. También contribuye al diagnóstico la realización del análisis molecular genético, siendo ambas pruebas complementarias específicas de dicha enfermedad.
]]> El tratamiento consiste en eliminar los tóxicos y evitar su producción. Suprimir el aporte proteico y favorecer el anabolismo con soluciones glucosadas y lípidos. Posteriormente se inicia el aporte proteico de forma escalonada con soluciones de aminoácidos. Se debe mantener el aporte proteico por encima del 25 % del basal. Tras normalizar el amonio se debe aportar proteínas sin aminoácidos precursores (valina, isoleucina, metionina y treonina). Además, se debe corregir la acidosis con bicarbonato y la hiperamonemia con fenilbutirato o benzoato sódico en caso de ser leve o moderada y con carbamil glutamato en casos severos. Es importante eliminar las toxinas mediante diuresis forzada, hemodiálisis o con diálisis peritoneal. El tratamiento corresponde a carnitina a dosis de 300 mg/día y vitamina B12 (1mg/día) en los casos que responden a ella5-6.Se recomienda realizar el control de los pacientes de forma mensual a los menores de 2 años, trimestral a los mayores de 2 años y tras una crisis de descompensación se debe revisar a los 15 días, tras la modificación de la dieta.
En cuanto a la metoclopramida, es un procinético intestinal y un bloqueador dopaminérgico y entre sus efectos adversos está descrita la disquinesia tardía. El tiempo tras el cual aparecen los síntomas es controvertido pero debe ser al menos 3 meses de tratamiento. Entre las formas más inusuales de la disquinesia tardía se encuentra el Tourette tardío. Existe un aumento de la predisposición de desarrollar la disquinesia tardía según sexo, edad y patologías subyacentes (pacientes con daño neurológico previo, enfermedad psiquiátrica de base, diabetes mellitus), reacción adversa a antipsicóticos, uso de anticolinérgicos centrales, tabaco, terapia electroconvulsiva y antecedentes familiares de disquinesia tardía. El pronóstico puede mejorar al ceder el tratamiento causante pero rara vez desaparece7.
En conclusión, la acidemia metilmalónica es una enfermedad de difícil diagnóstico dada su clínica inespecífica y la rareza de la patología. Es importante que el personal implicado en el manejo de dicha patología cuente con los conocimientos necesarios para la realización del diagnóstico diferencial. Las complicaciones que dicha acidemia, en caso de retraso de su diagnóstico, puede ocasionar incluyen desde el daño neurológico (por síndrome metabólico que afecta al tronco del encéfalo) hasta desarrollar una insuficiencia renal terminal, coma y muerte. Por tanto, es evidente la importancia de su diagnóstico precoz. En el caso de recién nacidos pueden no sobrevivir al primer episodio de síntomas y no llegan a cumplir los dos meses de edad.
Por otra parte, se debe tener en cuenta que existen fármacos que pueden inducir efectos adversos cuya presencia puede ser más probable en función de la enfermedad de base y de otros criterios que debemos considerar. Sabiendo que la AMM en sí puede provocar trastornos del movimiento, se debe tener en cuenta la medicación que se usa y el tiempo administrado.
Es importante que el paciente crezca con una buena base diabetológica y que conozca las complicaciones desde edad muy temprana. Su adecuado manejo en el ámbito de atención primaria corresponde tanto al médico como a enfermería.
Agradecimientos
Agradecemos la colaboración de Patricia Buller Viqueira, licenciada en filología inglesa, por la traducción realizada.
]]> Bibliografía
1. Mesa O, Ruiz M, García V, León J, López S, Solís C, et al. Aciduria metilmalónica con homocistinuria: una causa muy poco frecuente de fallo renal en el período neonatal. Nefrología. 2014; 34:539-40. [ Links ]
2. Orpha.net. (Internet) 2012 March. (consultado 22 de Octubre 2015). The portal for rare diseases and orphan drugs. Disponible en: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=293355. [ Links ]
3. Méndez ST, Vela-Amieva M, Velázquez-Arellano A, Ibarra I, Flores ME. Análisis de mutaciones en el gen de la metilmalonilCoA mutasa en diez pacientes mexicanos con academia metilmalónica aislada. Rev Inves Clin. 2012; 64:255-61. [ Links ]
4. Matusai SM, Mahoney MJ, Rosenberg LE. The Natural History of the Inherited Methylmalonic Acidemias. N Engl J Med. 1983; 308:857-61. [ Links ]
5. Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. [ Links ]
6. Ozand P, Rashed MS, Gascon GG, Youssef NG, Harfi H, Rahbeeni Z, al Garawi S, al Ageel A: Unusual presentations of propionic acidemia. Brain Dev.1994;16:46-57. [ Links ]
7. Venegas P, Millán María E, Miranda M. Disquinesia tardía. Rev Chil Neuro-Psiquiat. 2003; 41(2):131-8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Eva Buller Viqueira.
Avda. de Andalucía no 8. ]]>
C.P. 11150. Vejer de la Frontera.
Cádiz (España).
Correo electrónico: miji_77@yahoo.com
Recibido el 21 de diciembre de 2015.
Aceptado para su publicación el 3 de mayo de 2016.