Enfermedad de Niemann-Pick: un enfoque global
Niemann-Pick disease: a global approach
Diana Villamandos García y Alejandro Santos-Lozano
RESUMEN
]]> Niemann-Pick (NP) es una enfermedad lisosomal transmitida por herencia autosómica recesiva que se caracteriza por la acumulación de esfingomielina, colesterol y otros lípidos en diferentes órganos causando alteraciones celulares y viscerales. La enfermedad se puede subdividir en cuatro tipos (A, B ,C y D) y los síntomas más comunes son la visceromegalia y la afectación neuronal. El tratamiento de NP es principalmente paliativo y busca demorar los síntomas y, con ello, la muerte. Sin embargo, la investigación de la enfermedad continúa progresando para conseguir un diagnóstico temprano y un tratamiento que consiga disminuir el avance de la enfermedad.Palabras clave: enfermedades raras; enfermedades de depósito lisosomal; enfermedades de depósito lisosomal; sistema nervioso.
ABSTRACT
Niemann-Pick disease (NP) is a lysosomal disease that is transmitted by autosomal recessive inheritance. NP is characterized by the accumulation of sphingomyelin, cholesterol and other lipids in different organs causing cellular and visceral disorders. The disease can be subdivided into four types (A, B, C and D) and the most common symptoms are visceromegaly and neuronal involvement. NP treatment is mainly palliative and its principal aim is to delay the symptoms and to delay the death. However, the investigation of NP continues trying to get an accurate diagnosis at an early age and a treatment that could reduce the progression of the disease.
Key words: Rare Diseases; Lysosomal Storage Diseases; Lysosomal Storage Diseases; Nevous System.
En el año 1914, un pediatra alemán llamado Albert Niemann describió una serie de síntomas de lo que hasta entonces era una enfermedad desconocida (2, 3). Sus hallazgos provenían de una paciente judía de 18 años que manifestaba esplecnomegalia e hinchazón y pigmentación de la piel. La paciente murió a las pocas semanas. Niemann estaba seguro de que dichos síntomas eran característicos de la enfermedad de Gaucher, sin embargo, la muerte temprana de la paciente le hizo pensar que quizás fuera una variación de dicha enfermedad. No obstante, no fue hasta 1927 cuando un patólogo alemán, Ludwig Pick, analizó casos de niños con síntomas parecidos a los especificados por Niemann, encontrando semejanzas entre ellos (2). Es entonces cuando esta afección fue diferenciada de la enfermedad de Gaucher, acuñando el nombre de enfermedad de Niemann-Pick (NP), en honor a ambos facultativos alemanes (2-4).
Existen numerosas patologías relacionadas con anomalías en los lisosomas, son las llamadas "enfermedades lisosomales", entre las cuales se encuentra la enfermedad a estudio, NP (3, 5). El concepto de "enfermedad de almacenamiento lisosomal" fue introducido por HG. Hers en 1963 y, hoy en día, se conocen más de 40 enfermedades incluidas en esta denominación (3).
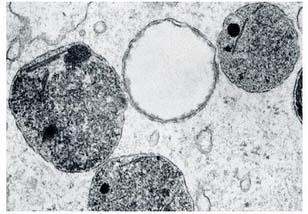
Los lisosomas son orgánulos celulares cuya función es la degradación de diferentes moléculas. Están formados una membrana y una cavidad o lumen (figura 1) donde se lleva a cabo el catabolismo de sustancias gracias a enzimas que presentan un pH ácido. Las enzimas lisosomales más abundantes son las hidrolasas ácidas, las cuales son activas a un pH ~ 5. Si existe un defecto genético en cualquiera de las estructuras que forman el lisosoma, se producirá un funcionamiento anómalo de este orgánulo debido a la incapacidad de degradar las macromoléculas. Como resultado, éstas se acumularán en el lisosoma formando inclusiones intracelulares (3, 5, 6).
]]> Figura 1. Lisosomas.
Niemann-Pick
NP engloba un conjunto de anomalías hereditarias autosómicas recesivas (3) y, se caracteriza por un acúmulo de diferentes lípidos, siendo los más abundantes la esfingomielina y el colesterol, en diferentes órganos y estructuras como el hígado, los nervios, el bazo, el cerebro y, en casos graves, los pulmones (1, 3, 7). La esfingomielina es una sustancia importante en las membranas celulares y constituye uno de los principales fosfolípidos de las vainas de mielina (3).
En 1961, Crocker clasificó la enfermedad en cuatro tipos en función de los órganos a los que afecta y la edad a la que aparecen los síntomas (3, 4, 8): NP tipo A (NPA), NP tipo B (NPB), NP tipo C (NPC) y tipo D (NPD).
En el año 1966, Brady demostró que los pacientes con NPA tenían en sus tejidos una deficiencia de la enzima esfingomielasa ácida (ASM) (9, 10), enzima que se encuentra en los lisosomas y que, en circunstancias normales, degrada la esfingomielina presente en las células (10). Este hecho se extendió al grupo NPB, pero no a NPC ni NPD (4, 10). Más adelante, se descubrió que el tipo NPC era causado por un fallo en el transporte de colesterol en el interior de la célula (4). Debido a estas consideraciones, los cuatro tipos de NP se asocian en dos grandes categorías (1, 3):
* Tipo I: NPA y NPB. Causados por el déficit de ASM. Si no hay suficiente ASM, la esfingomielina se acumula en las células ocasionando un mal funcionamiento de los diferentes órganos (3).
Este déficit primario de ASM es consecuencia de una mutación en el gen de la ASM (SMPD1) que está localizado en la sub-banda 1 o 4, de la banda 5, de la región 1 del brazo corto del cromosoma 11 (11p15.1-15.4) (3, 7).
NPA se caracteriza por un comienzo neonatal y por una muerte temprana en torno a los 2-3 años de edad (7). Sin embargo, el tipo NPB tiene una edad de diagnóstico variable aunque comúnmente suele comenzar en la infancia tardía (> 6 años) o la edad adulta (7, 10). En muchos casos, los pacientes con NPB logran vivir la adolescencia e incluso pueden llegar a vivir la edad adulta (10).
]]> En conjunto, los tipos NPA y NPB tienen una incidencia aproximada de 1 caso entre 250.000 nacidos vivos (7).* Tipo II: NPC y NPD. Esta categoría se caracteriza por un defecto en el transporte de lipoproteínas de baja densidad (LDL) derivadas del colesterol que, como consecuencia, provoca la acumulación de colesterol libre (no esterificado) y de glucoesfingolípidos en los lisosomas (1, 11, 12). Además, cursa de forma secundaria con un descenso de la ASM y un consiguiente acúmulo de esfingomielina en las células (3, 13). Estos lípidos se acumulan en numerosos órganos y tejidos, especialmente en el hígado, el bazo y el cerebro (14).
NPC es consecuencia de una mutación de los genes NPC1 o NPC2 (15) que, en circunstancias normales, codifican proteínas responsables del transporte lipídico intracelular (16). Las mutaciones en estos genes se manifiestan con una incidencia de 1 caso entre 120.000-150.000 nacimientos (3, 17):
* El gen NPC1 se encuentra localizado en el cromosoma 18 (18q11.2) y es el responsable de un 95% de las mutaciones que causan NPC (18).
* El gen NPC2, por su parte, tiene una presentación poco común (4%) y se puede localizar en el cromosoma 14 (14q24.3). (1, 18)
Los síntomas pueden aparecer a cualquier edad pero afectan sobre todo a niños y adolescentes. Podemos establecer 3 grupos dentro de NPC según la edad de diagnóstico (14, 19):
* NPC con inicio neonatal e infancia temprana: menor de 6 años
* NPC con inicio en infancia tardía: entre 6 y 11 años
* NPC con inicio juvenil y edad adulta: 12 años o más
NPD deriva también de una mutación en el gen NPC1 y, se puede considerar como una variante bioquímica y clínicamente casi idéntica al NPC (15). Por ello se pueden incluir en un único tipo, el NPC, ya que el NPD constituye una variante del mismo (1).
]]>Signos y síntomas
Además de las distintas características definitorias de cada tipo de NP, se puede también distinguir cada uno de ellos mediante los signos y síntomas que manifiestan los pacientes:
* NPA: son comunes las manifestaciones de hepatoesplecnomegalia, neurodegeneración, dificultades en la deglución y vómitos. Además, aparecen en la retina unas manchas características de color rojo cereza (3, 7).
* NPB: Es frecuente la aparición de hepatoesplecnomegalia y, tal y como ocurría con el tipo NPA, son usuales las manchas color rojo cereza en la retina de los enfermos (3, 7). Muchos pacientes con NPB manifiestan acumulación de esfingomielina en la médula ósea y en los pulmones, lo cual provoca infecciones bronquiales, hipoxia crónica y la muerte en casos severos (3, 7, 10).
Los pacientes con casos graves de NPB pueden presentar cirrosis hepática e hipertensión portal, sin embargo, no presentan afectación neurológica (1, 7, 12).
* NPC: Cuánto menor es la edad de aparición de los síntomas, más rápida es la degeneración neuronal, manifestándose principalmente en ataxia, distonía, disfagia, disartria, convulsiones, cataplexia y deterioro cognitivo (14, 15, 20).
Sin embargo, los síntomas de NPC son muy variables, no son específicos de la enfermedad y, se originan y desarrollan en distintos periodos de tiempo. Esto deriva en un diagnóstico difícil y, en muchas ocasiones, erróneo (15).
La edad de inicio de NPC, la sintomatología y el desarrollo de la enfermedad, así como la esperanza de vida, varían de unos pacientes a otros (14, 19). El inicio neonatal e infancia temprana de la enfermedad normalmente se manifiesta con ictericia neonatal transitoria, disfunción y fallo hepático y visceromegalia (comúnmente hepatomegalia y/o esplecnomegalia). En cuanto al inicio de infancia tardía, este se caracteriza por un rápido deterioro neurológico y por organomegalia variable. Finalmente, el comienzo juvenil o en edad adulta, generalmente se presenta con ataxia, patologías motoras y demencia o mayor proporción de problemas psiquiátricos (16, 17, 19).
Además aparecen, en tiempo variable, síntomas neurológicos y movimientos sacádicos oculares. Estos pacientes pueden desarrollar problemas en el trabajo/colegio y, en etapas maduras de la enfermedad, pueden manifestarse problemas conductuales, crisis epilépticas o problemas en el movimiento (11, 15, 21).
]]> Generalmente, cuando antes se manifiesten los síntomas, el desarrollo de la enfermedad es mayor y la muerte prematura ocurre antes en el tiempo (14).
Diagnóstico
El diagnóstico de la enfermedad de NP radica en el conocimiento de los síntomas y en la sospecha clínica. En los lactantes y niños de infancia temprana es importante la asociación de la visceromegalia como un posible síntoma de la enfermedad (12).
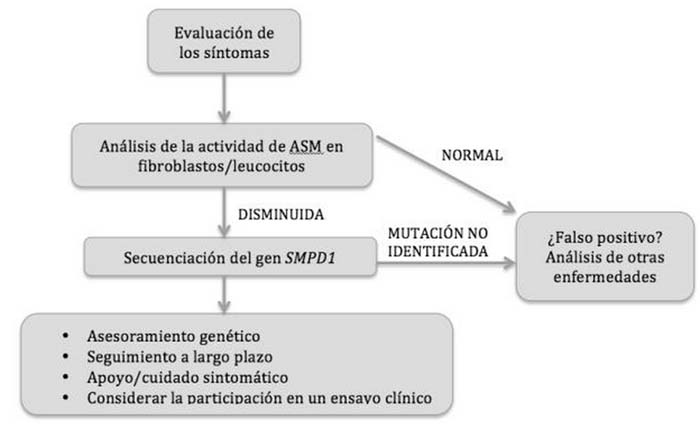
* Tipos NPA y NPB (figura 2): el diagnóstico primario se realiza mediante la evaluación de la actividad de la ASM en leucocitos y fibroblastos, la medición de los niveles de transaminasas y de bilirrubina y el perfil lipídico (7). Se ha demostrado que la actividad de la ASM en el tipo NPA de la enfermedad es <5% de lo normal, por lo que los niveles de esfingomielina son muy elevados. En cambio, en el tipo NPB de la enfermedad la actividad de la ASM es mayor, constituyendo un 2-10% de su actividad normal (7, 13). Si la actividad de la ASM es baja, se procederá a la secuenciación del gen SMPD1 y el análisis de su posible mutación (7).
Figura 2. Algoritmo diagnóstico para NPA y NPB (7)
Se han revelado altos niveles de esfingomielina en el tipo NPA y valores normales en NPB en estudios realizados en cerebros de personas fallecidas que padecían la enfermedad de NP. Pero, sin duda, el hallazgo más significativo es la aparición de células espumosas (figura 3) en tejidos del hígado, pulmón o médula ósea (7). Las células espumosas son macrófagos cargados de lípidos que se forman a partir de la captación masiva de LDL y la acumulación en su citoplasma de ésteres de colesterol (22). La presencia de dichas células en los tejidos hace que se manifiesten los síntomas descritos anteriormente: visceromegalia, afectación pulmonar o disfunción de la médula ósea (7).
]]>Figura 3. Células espumosas en aspirado de médula ósea (1) 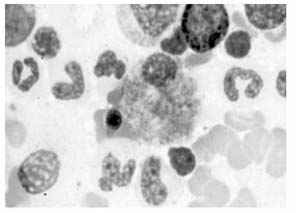
* Tipo NPC (figura 4): el diagnóstico del tipo NPC de la enfermedad radica en la demostración de la acumulación de colesterol no esterificado en los lisosomas o la demostración de la disminución del colesterol esterificado en el citoplasma (11). La demostración de la acumulación lipídica de la enfermedad se centra en la técnica de tinción de filipina en células vivas, usualmente en fibroblastos de la piel (11). Esta prueba de laboratorio consiste en la utilización de un antibiótico sintético (filipina) para diagnosticar la presencia de colesterol esterificado LDL en cultivos de fibroblastos. Dichas células se exponen a la filipina que, en contacto con el LDL presente en ellos producirá una fluorescencia visible al microscopio (15, 23).
Figura 4. Algoritmo diagnóstico para NPC (15). 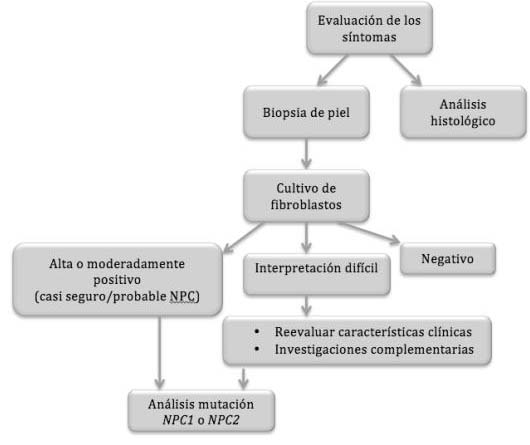
El análisis histológico puede determinar la presencia de células espumosas y/o histocitos azul-mar en la médula ósea, hígado, bazo, pulmones o nódulos linfáticos. Este hallazgo característico, aunque no específico, sirve para la aproximación al diagnóstico de NPC (11, 15).
]]> Si el test con filipina muestra algún indicio de positividad se procederá a la prueba genética en la que se determinará si existe mutación en los genes NPC1 o NPC2 y, por tanto, si el paciente presenta o no NPC (15).
Tratamiento
Por el momento no existe cura capaz de acabar con la enfermedad de NP debido a las preguntas que aún quedan por responder sobre la causa que provoca esta alteración. Sin embargo, son muchos los ensayos clínicos que se han llevado o se están llevando a cabo para dar con un tratamiento que consiga, al menos, retrasar los síntomas.
En los tipos NPA y NPB la investigación se centra en el trasplante de células hematopoyéticas y en el reemplazamiento enzimático (7, 12). La mayor parte de los ensayos clínicos sobre el tratamiento de la enfermedad están basados en el tratamiento de NPC, aunque tampoco existe un tratamiento capaz de alterar el ritmo de la enfermedad (14, 24). El único tratamiento disponible en Europa para NPC es Miglustat. Se trata de un fármaco capaz de inhibir la enzima responsable de la catalización del primer paso para la síntesis de glucoesfingolípidos (14, 17, 24, 25). Su uso se limita al tratamiento de las manifestaciones neurológicas en pacientes tanto adultos como pediátricos de NPC (14).
No existe cura capaz de acabar con la enfermedad de NP y su tratamiento es un tema aún por desarrollar. Sin embargo, el objetivo actual es desarrollar tratamientos con el fin de reducir al mínimo los síntomas y retrasar al máximo la degeneración neuronal. Para ello, se llevan a cabo ensayos clínicos que llevan a la práctica fármacos aún en estudio para comprobar sus ventajas e inconvenientes.
Conclusión
La enfermedad NP, en sus cuatro subtipos, es una enfermedad poco común con signos y síntomas variados que dificultan su diagnóstico. El tratamiento del que se benefician los pacientes con esta enfermedad está destinado principalmente a paliar los síntomas y a ralentizar el avance de la enfermedad en la medida de lo posible.
Sin embargo y, pese a que aún quedan muchas preguntas sin respuesta, la investigación de la enfermedad de NP, así como de su tratamiento, progresa continuamente dando paso a nuevos avances que favorecen el diagnóstico en edades tempranas.
]]>Referencias bibliográficas
1. Guadalupe J-T, Fabiola O-P, David I-G. (2012). Enfermedad de Niemann-Pick tipo C. Revista Mexicana de Neurociencia Septiembre-Octubre.13(5):281-5. [ Links ]
2. Abel EL. (2001). Jewish Genetic Disorders. A Layman's Guide. North Carolina, USA: McFarland & Company. Available from: http://books.google.es/books?id=yUNKaRAVZvMC&printsec=frontcover&dq=jewish+genetic+disorders&hl=es&sa=X&ei=9bgVU6_rIYG2yAPtwYGQBQ&ved=0CDAQ6AEwAA-v=onepage&q&f=false. [ Links ]
3. Stern G. (2014). Niemann-Pick's and Gaucher's diseases. Parkinsonism & Related Disorders. 20:S143-S6. [ Links ]
4. Vanier M. (2010). Niemann-Pick disease type C. Orphanet Journal of Rare Diseases. 5(1):16. PubMed PMID: doi:10.1186/1750-1172-5-16. [ Links ]
]]>5. Boya P, Pérez-Sala D, Stamatakis K. (2010). Use of a protein sequence of localisation and endolysosomal degradation. [ Links ]
6. Cooper GM. (2000). The Cell: A Molecular Approach. Sunderland (MA): Sinauer Associates. 2nd. Available from: http://www.ncbi.nlm.nih.gov/books/NBK9953/. [ Links ]
7. Wang RY, Bodamer OA, Watson MS, Wilcox WR. (2011). Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genetics in medicine: official journal of the American College of Medical Genetics. 13(5):457-84. PubMed PMID: 21502868. Epub 2011/04/20. eng. [ Links ]
8. Crocker AC. (1961). The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. Journal of neurochemistry. 7:69-80. PubMed PMID: 13696518. Epub 1961/04/01. eng. [ Links ]
9. Brady RO, Kanfer JN, Mock MB, Fredrickson DS. (1966). The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proceedings of the National Academy of Sciences of the United States of America. 55(2):366-9. PubMed PMID: 5220952. Pubmed Central PMCID: PMC224150. Epub 1966/02/01. eng. [ Links ]
]]>10. Thurberg BL, Wasserstein MP, Schiano T, O'Brien F, Richards S, Cox GF, et al. (2012). Liver and skin histopathology in adults with acid sphingomyelinase deficiency (Niemann-Pick disease type B). The American journal of surgical pathology. 36(8):1234-46. PubMed PMID: 22613999. Pubmed Central PMCID: 3396757. [ Links ]
11. Marfa MP. (2012). Enfermedad de Niemann-Pick C. Barcelona: Publicaciones Permanyer. [ Links ]
12. McGovern MM, Wasserstein MP, Giugliani R, Bembi B, Vanier MT, Mengel E, et al. (2008). A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 122(2):e341-9. PubMed PMID: 18625664. Pubmed Central PMCID: PMC2692309. Epub 2008/07/16. eng. [ Links ]
13. Salegio EA, Samaranch L, Jenkins RW, Clarke CJ, Lamarre C, Beyer J, et al. (2012). Safety study of adeno-associated virus serotype 2-mediated human acid sphingomyelinase expression in the nonhuman primate brain. Human gene therapy.;23(8):891-902. PubMed PMID: 22574943. Pubmed Central PMCID: PMC3413900. Epub 2012/05/12. eng. [ Links ]
14. Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. (2010). Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Molecular genetics and metabolism. 99(4):351-7. PubMed PMID: 20045366. [ Links ]
]]>15. Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, et al. (2009). Recommendations on the diagnosis and management of Niemann-Pick disease type C. Molecular genetics and metabolism. 98(1-2):152-65. PubMed PMID: 19647672. [ Links ]
16. Héron Bnd, Valayannopoulos V, Baruteau J, Chabrol B, Ogier Hln, Latour P, et al. (2012). Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet Journal of rare Diseases. [ Links ]
17. Wraith JE, Imrie J. (2009). New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. Therapeutics and clinical risk management. 5:877-87. PubMed PMID: 19956552. Pubmed Central PMCID: PMC2781062. Epub 2009/12/04. eng. [ Links ]
18. Science TWIo. The GeneCards Human Gene Database (Internet)1996-2014 (updated 23 Oct 2013; cited 2014 11 Febrero). Available from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=NPC2&search=NPC2. [ Links ]
19. Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, et al. (2009). Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Molecular genetics and metabolism. 98(3):243-9. PubMed PMID: 19656703. [ Links ]
]]>20. Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B, et al. (2009). Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Molecular genetics and metabolism. 98(3):250-4. PubMed PMID: 19616462. [ Links ]
21. Abel LA, Bowman EA, Velakoulis D, Fahey MC, Desmond P, Macfarlane MD, et al. (2012). Saccadic eye movement characteristics in adult Niemann-Pick Type C disease: relationships with disease severity and brain structural measures. PloS one. 7(11):e50947. PubMed PMID: 23226429. Pubmed Central PMCID: PMC3511378. Epub 2012/12/12. eng. [ Links ]
22. Valledor AF, Lloberas J, Celada A. (2001). Macrophage Foam Cells. eLS: John Wiley & Sons, Ltd. [ Links ]
23. Ledvinova J, Elleder M. (1993). Filipin test for diagnosis of Niemann-Pick disease type C. Sbornik lekarsky. 94(2):137-43. PubMed PMID: 7992006. Epub 1993/01/01. eng. [ Links ]
24. Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. (2010). Long-term miglustat therapy in children with Niemann-Pick disease type C. Journal of child neurology. 25(3):300-5. PubMed PMID: 19822772. [ Links ]
]]>25. Patterson MC VD, Prady H, Abel L, Wraith JE. (2007). Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. [ Links ]
26. Yagyu H, Nagashima S, Takahashi M, Miyamoto M, Okada K, Osuga J, et al. (2012). Ezetimibe, an inhibitor of Niemann-Pick C1-like 1 protein, decreases cholesteryl ester transfer protein in type 2 diabetes mellitus. Endocr J. 28;59(12):1077-84. PubMed PMID: 22850130. Epub 2012/08/02. eng. [ Links ]
27. Takase H, Dohi Y, Okado T, Hashimoto T, Goto Y, Kimura G. (2012). Effects of ezetimibe on visceral fat in the metabolic syndrome: a randomised controlled study. European journal of clinical investigation. 42(12):1287-94. PubMed PMID: 23033884. [ Links ]
]]>