










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Internet)
versión On-line ISSN 1698-6946
Med. oral patol. oral cir.bucal (Internet) vol.11 no.3 may./jun. 2006
Síndrome de Cockayne: Informe de un caso. Revisión de la literatura
Cockaynes Syndrome: A case report. Literature review
María de la Luz Arenas Sordo 1,4, Edgar Hernández Zamora 1, Luis Alberto Montoya Pérez 2,
Beatriz Catalina Aldape Barrios 3
(1) Servicio de Genética, Instituto Nacional de Rehabilitación Secretaría de Salud
(2) Pasante de Odontología, Servicio Social, Universidad Nacional Autónoma
(3) Servicio de Patología Bucal, Estudios de Postgrado Universidad Nacional Autónoma
(4) Académico de la Universidad Nacional Autónoma. México
Dirección para correspondencia
RESUMEN
El Síndrome de Cockayne (CS) es un desorden genético con un patrón de herencia autosómico recesivo que fue descrito por primera vez en 1936 por Cockayne. Los pacientes con este síndrome presentan detención del crecimiento, talla baja, envejecimiento prematuro, anormalidades neurológicas, fotosensibilidad, retraso en la erupción de los dientes primarios, ausencia congénita de dientes permanentes, macrodoncia parcial, atrofia de los procesos alveolares y caries dental. Puede ser causado por mutación en dos genes, el CKN1 (ERCC8) y el ERCC6, localizados en los cromosomas 5 y 10 respectivamente; originando dos tipos: CS-A que tienen mutación en ERCC8 y CS-B con mutación en ERCC6, este último provoca sensibilidad a la luz ultravioleta, secundaria a una deficiencia en la reparación de DNA.También se ha asociado el síndrome a mutaciones de los genes XPB, XPD y XPG.
En el presente reporte se informa de un paciente de 9 años con cuatro meses de edad. En la exploración física se registró talla de 94 cm, peso de 8.6 kg y perímetro cefálico de 42 cm. TA 120/80. Hábito caquéctico, problemas posturales con encorvamiento, así como microcefalia, cara ovalada, ojos hundidos, nariz delgada y afilada, falta de grasa en la cara, más notorio en el tercio medio y orejas grandes que le confieren una apariencia de "pajarito". Se observa marcada fotosensibilidad en toda la piel expuesta al sol. Presenta retraso psicomotor y mental.
Intrabucalmente se aprecia higiene deficiente, gingivitis, caries cervical, hipoplasia del esmalte, mala posición dentaria de los incisivos laterales superiores e inferiores, y macrodoncia de los dientes centrales superiores, el izquierdo presenta una lesión por caries. Radiográficamente se observa ausencia congénita de los dientes 14, 23, 24 e hipoplasia mandibular.
El objetivo de este trabajo es dar a conocer a la comunidad odontológica las características del síndrome de Cockayne a través de un caso clínico.
Palabras clave: Envejecimiento prematuro, fotosensibilidad, gen CKN1, gen CSA, gen CSB, gen ERCC6, gen ERCC8, gen XPB, gen XPD, gen XPG, macrodoncia, oligodoncia, retraso en el crecimiento y desarrollo, síndrome de Cockayne.
ABSTRACT
Cockaynes syndrome is a genetic disorder with a recessive autosomal inheritance, described first by Cockayne in 1936. Patients with this syndrome present failure to thrive, short stature, premature aging, neurological alterations, photosensitivity, delayed eruption of the primary teeth, congenitally absent of some permanent teeth, partial macrodontia, atrophy of the alveolar process and caries. It could be caused by two gene mutations, CNK1 (ERCC8) and ERCC6, located on the 5 and 10 chromosomes respectively, causing two variations of Cockaynes syndrome, CS-A, secondary to a ERCC8 mutation and CS-B with ERCC6 mutation, the last one causes hypersensitivity to the ultraviolet light secondary to a DNA repair defect. The syndrome is also associated with mutations of the XPB, XPD and XPG genes.
In this report we present a 9 year and 4 month old patient. He had a height of 94 cm, weight of 8.6 Kg, head circumference of 42 cm. and blood pressure of 120/80. Cachectic habitus, kyphosis, microcephaly, oval face, sunken eyes, a thin and beaklike nose, lack of subcutaneous facial fat (especially in the middle of the face), and large ears give the patient a birdlike appearance. It is notorious the photosensitivity in all the sun-exposed skin. The patient also displays delayed psychomotor skills and mental retardation.
In the oral cavity we found deficient hygiene, gingivitis, cervical caries, enamel hipoplasia, abnormal position of the upper and inferior lateral incisors, macrodontia of the upper central teeth, the left one presented a caries. In the x-ray we observed congenital absence of 14, 23 and 24 teeth and mandibular hipoplasia.
The aim of this review is to show the dentistry community the characteristics of the Cockaynes syndrome by means of a clinical case.
Key words: Premature aging, photosensitivity, CKN1 gene, CSA gene, CSB gene, ERCC6 gene, ERCC8 gene, XPB gene, XPD gene, XPG gene, macrodontia, oligodontia, growth and development retardation, Cockaynes syndrome.
Introducción
Existen por lo menos quince desórdenes humanos causados por defectos en la reparación del ADN (1). Uno de estos es el Síndrome de Cockayne (CS), descrito por primera vez en 1936 por Cockayne (2,3).Este síndrome se presenta aproximadamente en 1 de cada 100 000 nacidos vivos y puede ser causado por mutación en dos genes diferentes, el CKN1 o ERCC8 (Excision-Repair Cross-Complementing, Group 8), y el ERCC6 (Excision-Repair Cross-Complementing, Group 6), localizados en el cromosoma 5 y 10q11 respectivamente; lo que origina dos síndromes clínicos: CS-A para el CKN1 y CS-B para ERCC6.También se ha asociado con mutaciones de los genes XPB (Xeroderma pigmentoso B), XPD (Xeroderma pigmentoso D) y XPG (Xeroderma pigmentoso G) (1,3-6).
Las manifestaciones clínicas del CS son detención del crecimiento, envejecimiento prematuro, talla baja, aspecto caquéctico, longitud desproporcionada de las extremidades, cifosis, microcefalia y cabello escaso y disperso. Falta de grasa en la cara, particularmente en las mejillas, prominencia de huesos faciales, microcefalia, ojos hundidos, nariz en pico y orejas grandes, que dan la apariencia de "pajarito" (1,2,5-7). Presenta asimismo, marcada fotosensibilidad a la luz UV sin que, aparentemente, se incremente el riesgo de desarrollar cáncer de piel (6-9).
Los pacientes con este síndrome sufren de retraso mental y del desarrollo psicomotor, que se reflejan en la incapacidad para hablar y caminar (2,5-8,10,11), aunque se han informado casos en los que no se encuentra alterada la inteligencia (7). En algunos pacientes se observan alteraciones oftalmológicas como retinopatía pigmentaria, degeneración retiniana, cataratas y disminución en la secreción lagrimal, así como pérdida de la audición. Algunas otras complicaciones que pueden presentarse son hepatoesplenomegalia, problemas renales e hipertensión arterial (3,5,7).
Las manifestaciones bucales que se presentan son retraso en la erupción de la primera dentición, que varía desde 6 meses a 4 años según los casos informados, ausencia congénita de varios dientes permanentes (oligodoncia), macrodoncia parcial principalmente de los incisivos centrales, hipoplasia dental, raíces cortas, mayor incidencia de caries, paladar profundo, atrofia del proceso alveolar, prognatismo mandibular e hipoplasia condilar (3,5,7,10,11).
Caso clínico
Paciente masculino de 9 años con cuatro meses de edad, sin antecedentes heredofamiliares de importancia, consanguinidad y endogamia negadas. Fue producto de la segunda gestación, con duración del embarazo de 36 semanas. Al nacer pesó 2.900 kg, presentó cataratas congénitas completas, que fueron tratadas quirúrgicamente. Su desarrollo psicomotor fue muy retrasado. A los 3 años su crecimiento y desarrollo se estancó bruscamente. En la exploración física se observó talla de 94 cm y peso de 8.6kg, perímetro cefálico de 42 cm, el valor de su presión arterial fue de 120/80 mm/Hg. A la exploración física se encontró: hábito caquéctico, problemas posturales con encorvamiento, la cara de forma ovalada, ojos hundidos, nariz delgada y afilada, falta de grasa en las mejillas, orejas grandes, lo que le confiere en conjunto una apariencia de "pajarito". Se observa marcada fotosensibilidad principalmente en la cara. También es notorio el retraso psicomotor y mental. No camina y es incapaz de mantenerse en bipedestación. Fig.1.
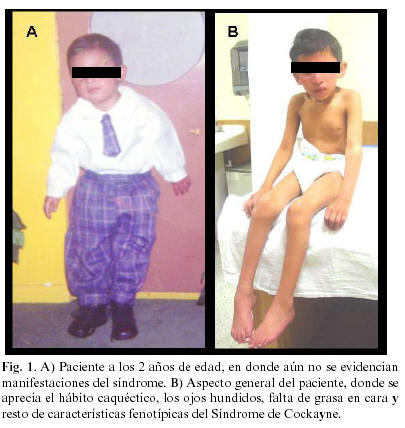
En la exploración bucal encontramos a la mucosa de consistencia normal, el paladar estrecho de profundidad ligeramente aumentada, higiene deficiente y gingivitis. Los dientes erupcionados son 11, 12, 16, 21, 22, 26, 31, 32, 33, 34, 36, 41, 42, 43 y 46, parcialmente erupcionados están el 15 y el 25. De ellos el 12, 22, 32 y 42 están ligeramente girados, el 11 y 21 presentan macrodoncia, en el 21 se presenta caries a nivel del tercio incisal, el 16, 26, y 36 están hipoplásicos, los dientes 26 y 46 están obturados con amalgamas. Los incisivos inferiores están afectados por caries cervical. Radiográficamente se aprecian los gérmenes de los dientes 13, 17, 27, 35, 37, 44, 45, 47, y ausencia congénita de 14, 23 y 24. Los dientes erupcionados tienen raíces cortas. La mandíbula se observa hipoplásica en el cuerpo, ramas y cóndilos, es notoria la atrofia del hueso alveolar.
Discusión
Las características fenotípicas del nuestro paciente nos permiten claramente realizar el diagnóstico de síndrome de Cockayne. La posibilidad de correlacionarlo con el tipo de mutación presente en el gen CSB no es posible, aunque asumimos que ésta permitió la existencia de la proteína que codifica este gen, a pesar de no ser normal, ya que la ausencia de la misma provocaría un fenotipo mucho más grave (10).
Nuestro paciente presenta presión arterial (120/80) por arriba de lo óptimo (diastólica <80), dato relevante por lo que puede significar para la salud del mismo. Esta es una característica referida en la literatura en la que se debe prestar atención por su significado clínico (12).
La importancia del presente caso radica en que es un padecimiento poco frecuente y presenta manifestaciones bucales importantes (5,9). Consideramos, de tal manera, que la comunidad odontológica debe estar abierta a la búsqueda de signos y síntomas de patología, fuera de la cavidad bucal, que les permita llegar a un diagnóstico integral y poder conocer la evolución del padecimiento para realizar el tratamiento adecuado.
No se conoce cómo la proteína anormal del gen CSB produce las alteraciones estomatológicas, ni muchas otras (11), por lo que pueden considerarse como futuras líneas de investigación.
Bibliografía
1. Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003;85:1101-11. [ Links ]
2. Rawlinson SC, Webster VJ. Spinal anaesthesia for caesarean section in a patient with Cockayne syndrome. Int J Obstet Anesth 2003;12:297-9. [ Links ]
3. Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck. New York: Oxford University Press; 2001. p. 596-600. [ Links ]
4. Shiomi N, Kito S, Oyama M, Matsunaga T, Harada YN, Ikawa M, et al. Identification of the XPG region that causes the onset of Cockayne syndrome by using Xpg mutant mice generated by the cDNA-mediated knock-in method. Mol Cell Biol 2004;24:3712-9. [ Links ]
5. Schneider PE. Dental findings in a child whit Cockayne´s syndrome: Case reports. J Dent Child 1983;50:58-64. [ Links ]
6. Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst). 2002;1:913-27. [ Links ]
7. Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet 1992;42:68-84. [ Links ]
8. Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, et al. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15410-5. [ Links ]
9. Lee SK, Yu SL, Prakash L, Prakash S. Requirement of yeast RAD2, a homolog of human XPG gene, for efficient RNA polymerase II transcription. implications for Cockayne syndrome. Cell. 2002;109:823-34. [ Links ]
10. Spivak G. The many faces of Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15273-4. [ Links ]
11. Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology 2000;55:1442-9. [ Links ]
12. Williams B, Poulter NR, Brown MJ, Davis M, McInnes GT, Potter JF, et al. Guidelines for management of hypertension: report of the fourth working party of the British Hypertension Society, 2004-BHS IV. J Hum Hypertens 2004;18:139-85. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dra. María de la Luz Arenas Sordo
Centro Nacional de Rehabilitación S.S.
Servicio de Genética (Torre de Investigación tercer piso).
Calzada México-Xochimilco 289,
Col. Arenal de Guadalupe.
Delegación Tlalpan, C.P. 14389, México D.F.
E-mail: asgk@servidor.unam.mx
Recibido: 23-02-2005
Aceptado: 21-01-2006

