










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
versión impresa ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.10 no.5 nov./dic. 2005
Hereditary familial polyposis and Gardner's syndrome:Contribution of
the odontostomatology examination in its diagnosis
and a case description
Poliposis familiar hereditaria y síndrome de Gardner: Aportación de la exploración
odontoestomatológica a
su diagnóstico y descripción de un caso
Eduardo Chimenos-Küstner (1), Montserrat Pascual (2), Ignacio Blanco (3), Fernando Finestres (4 )
(1) Estomatólogo. Doctor en Medicina y Cirugía. Profesor Titular de Medicina Bucal,
Facultad de Odontología, Universidad de Barcelona
(2) Estomatóloga. Licenciada en Medicina y Cirugía. Área Básica de Salud Sant Roc (Badalona).
Profesora Asociada de la Facultad de Medicina de la Universidad Autónoma de Barcelona
(3) Oncólogo. Doctor en Medicina y Cirugía. Unidad de Consejo Genético, Servicio de Prevención y Control del Cáncer,
Instituto Catalán de Oncología. Profesor Asociado de Biología Celular y Anatomía Patológica,
Facultad de Medicina, Universidad de Barcelona
(4)Radiólogo y Estomatólogo. Doctor en Medicina y Cirugía. Profesor Asociado de Medicina Bucal,
Facultad de Odontología, Universidad de Barcelona
Address:
Dr. Eduardo Chimenos Küstner
Vía Augusta 124, 1º 3ª
08006 – Barcelona (España)
E-mail: 13598eck@comb.es
Received: 23-05-2004 Accepted: 20-11-2004
|
Chimenos-Küstner E, Pascual M, Blanco I, Finestres F. Hereditary familial polyposis and Gardner's syndrome:
Contribution of the odonto-stomatology examination in its diagnosis and a case description. Med Oral Patol Oral Cir Bucal 2005;10:402-9. |
|
ABSTRACT
Familial adenomatous polyposis (FAP) and its phenotype variant, Gardner's syndrome, constitute a rare autosomal dominant inherited disorder. They are characterised by the development, generally during the second and third decades of life, of multiple adenomatous polyps in the colon and rectum. These polyps have a high risk of subsequently becoming malignant, which normally occurs in the third and fourth decades of life. The phenotypical features of FAP can be very variable. As well as colorectal polyps, these individuals can present with extra-colonic symptoms, among which are particularly: gastro-duodenal polyps, dermoid and epidermoid cysts, desmoid tumours, congenital hypertrophy of the retinal pigment epithelium, disorders of the maxillary and skeletal bones and dental anomalies. In this paper the most important aspects of this syndrome are reviewed, showing an example based on a well documented clinical case. The importance of odonto-stomatological examinations should be pointed out, among others, as a means of reaching a presumptive diagnosis, whose confirmation is vital to the patient. Key words: Hereditary familial polyposis, Gardner's syndrome, osteomas, diagnostic markers. |
|
RESUMEN La poliposis adenomatosa familiar (PAF) y su variante fenotípica, el síndrome de Gardner, constituyen una infrecuente patología hereditaria autosómica dominante. Se caracterizan por el desarrollo, generalmente durante la segunda y tercera década de la vida, de múltiples pólipos adenomatosos en el colon y en el recto. Estos pólipos tienen un riesgo elevado de transformación maligna subsiguiente, cosa que suele ocurrir en las décadas tercera y cuarta de la vida. Las manifestaciones fenotípicas de la PAF pueden ser muy variadas. Así, además de los pólipos colorrectales, los individuos afectos pueden presentar manifestaciones extracolónicas, entre las que se destacan: pólipos gastroduodenales, quistes dermoides y epidermoides, tumores desmoides, hipertrofia congénita del epitelio pigmentario de la retina, alteraciones óseas en los maxilares y en el esqueleto y anomalías dentarias. En este trabajo se revisan los aspectos más importantes del complejo, mostrando un ejemplo del mismo en base a un caso clínico bien documentado. Cabe destacar la importancia de las exploraciones odontoestomatológicas, entre otras, como medio para alcanzar el diagnóstico de presunción, cuya confirmación es vital para el enfermo. Palabras clave: Poliposis familiar hereditaria, síndrome de Gardner, osteomas, marcadores diagnósticos. |
INTRODUCTION
Familial Adenomatous Polyposis (FAP) is a rare dominant autosomal hereditary disease, which is characterised by the development, generally during the second and third decade of life, of multiple adenomatous polyps (more than 100 in number) in the colon and rectum. These polyps have a high risk of subsequently becoming malignant, which normally occurs in the third and fourth decades of life. The phenotypical symptoms of FAP can be very variable. As well as colorectal polyps, the individual affected can present with extra-colonic symptoms, among which are highlighted: gastro-duodenal polyps, dermoid and epidermoid cysts, desmoid tumours, congenital hypertrophy of the retinal pigment epithelium (CHRPE), disorders of the maxillary and skeletal bones and dental anomalies (1). Gardner's syndrome is characterised by colorectal polyps, cutaneous epidermal cysts and mandibular and long bone osteomas. Gardner's syndrome is considered as a phenotypical variant of FAP. Nowadays the genetic basis of Familial Adenomatous Polyposis, and its different phenotypical variants, is known. FAP and Gardner's syndrome are due to germline mutations in the APC gene (Adenomatous Polyposis Coli gene), located in long arm of chromosome 5 (2). The phenotype-genotype correlation in FAP starts to be recognised, in such a way that determined phenotypical manifestations are related with mutations in specific areas of the APC gene. The extra-colonic symptoms in FAP (for example, osteomas, dermoid cysts etc.) are related with mutations located between the 1395 and 1578 codons (2). This phenotype-genotype correlation will be of vital importance when genome repair techniques (genetic therapy) are available. Nowadays it is used in determining the measures of selection most appropriate to each case. On occasions it is not possible to determine a pathogenic mutation in the APC gene (3).
The aim of this paper is to highlight the importance of early detection of this pathology, taking into account that the finding of lesions which settle in the maxillaries play an important diagnostic role.
CLINICAL MANIFESTATIONS
Osteomas. Osteomas are benign osteogenic lesions characterised by the slow proliferation of compact or medullary bone. They can be central, peripheral or extra-skeletal. The central osteomas coming from the endosteum, the peripherals of the periosteum and the extra-skeletal, develop in the soft tissue, such as muscle. The lesion can be in more than one bone or with more than one osteoma in a single bone. In the maxillofacial region, the periosteum type can appear externally as well as in the para-nasal sinuses. It is more common in the frontal and ethmoid sinuses than in the maxillary sinuses. Structurally the osteomas can be divided into three types, depending on whether they are composed of compact bone, medullary bone or of a combination of compact and medullary bone. Clinically, the peripheral osteoma are normally asymptomatic, but can produce tumefaction and cause asymmetry. From a radiography point of view, the lesion is defined as a well circumscribed radio-opacity. The computerised tomography is the best imaging technique for the diagnosis of an osteoma. Histologically, the osteomas are normally formed by trabeculas of bone laminated with fibroadipose tissue. In the case of enostosis, these consist of compact islets of mature laminated bone (4-6). Although osteomas in the facial bones and cranium are not common in the general population, in patients affected by Gardner's syndrome they are. Frequently they are large masses, multi-lobed, in the goniac region. Many of them can converge with adjacent osteomas. Some osteomas can adopt a drop shape, which appears to hang from the lower edge of the mandibular or condyle border. The osteomas which originate in the bone medulla mimic enostosis. Approximately 50% of cases present with three or more osteomas in the maxillaries, as well as other locations. A common location for this type of exostosis is the frontal bone. Osteomas originating from the endochondral bone are rare, even in this syndrome. When they are found in long bones such as the tibia or the femur, they adopt more the aspect of cortical thickening, which is a true osteoma. It is very important to take into account that the appearance of osteomas precede the other manifestations of this syndrome, intestinal polyposis included. It also has to be remembered that, with an incidence of 17% odontomas, supernumery teeth and impacted teeth can be found (4-8).
Intestinal polyposis. Familial adenomatous polyposis of the colon is the most frequent syndrome amongst the hereditary polyposes (1/8000). The majority of individuals have a family history of this pathology, but up to 30% of patients can present with a new dominant mutation (“de novo” mutation) and be the first member of the family affected. The polyposis normally develops after the osteomas. The majority appear during the second and third decades of life. Their transformation to malignancy is a constant feature, which is only a matter of time. In puberty, the rate of malignancy is 5% , increasing to 50% at 30 years and 100% in cases more than 50 years of age. Also, Gardner's syndrome is associated with polyps in any part of the digestive tract, which in turn can become malignant. The presence of malignant tumours has been described in other locations: carcinomas in the ampula of Vater, medullolastomas, carcinomas of the thyroid and hepatoblastomas (4,9,10). The prevalence of cancer in patients with symptomatic familial adenomatous polyposis varies between 47 and 67%. Records show that despite knowledge of the disease, 59 % of these patients die due to a metastatic extension of colorectal cancer. However, when it is studied in asymptomatic families that present with the FAP phenotype, the prevalence of malignant transformation is seen in only 2%. The impact that the asymptomatic detection can have on the survival is very noticeable. Patients detected in the symptomatic stage have a 5 year survival rate of 40% , in comparison with 93% in individuals identified during detection programs (11, 12). From a histological point of view, the polyps of this syndrome are adenomatous and are found mainly in the colon. However the small intestine, in particular the duodenum, can be involved in this process. Also gastric polyps (polyps of the fundic glands), hamartomatous in nature, are frequently observed. The adenomas are tubular, hairy or a combination of both. Although the adenomas of the colon, especially the hairy adenomas, can become malignant in the general population, it has to borne in mind that in patients with Gardner's syndrome this is inevitable and it generally happens earlier. If a colectomy is not performed, it will develop into an adenoma-carcinoma. These tumours can secrete variable quantities of mucin, but the prognosis depends more on the tumour extension on the intestinal wall, than the specific histological characteristics (4).
Epidermoid, dermoid and sebaceous cysts. Sebaceous cysts develop in approximately 60% of cases. On average, the number is 4, although some individuals develop 20 or more. The cysts are seen most frequently on the face, on the scalp, the arms and legs. They also normally appear before puberty and intestinal polyposis appears. The structure of its wall is analogous to the skin and normally contains organised materials such as fat, hairs, glands etc. It is surrounded by a narrow stratified squamous epithelium, a keratin producer (4,10).
Fibromas and fibromatosis in soft tissues. These soft tissue tumours, often called abdominal or extra-abdominal desmoids, are infiltrated fibrous masses, which are seen in 15 to 30% of cases. Such fibrous tumours are seen in only 5% of FAP cases, due to its low penetration in the SG-FAP gene. Some of these tumours appear de novo, others after surgical treatment (especially abdominal surgery) and others after extirpation of previous desmoid tumours. Tumours localised in the maxillofacial areas have been seen that infiltrate the masticatory and suprahyoid musculature (4, 10).
Congenital hypertrophy of the retinal pigment epithelium (CHRPE). Up to 75% of patients with familial adenomatous polyposis (FAP) present with congenital hypertrophy of the retinal pigment epithelium, which is easily detected by ophthalmoscopy. As it is not common for a normal individual to present with this type of lesion, its detection, along with some all ready mentioned clinical data, must include consideration of the possibility of Gardner's syndrome (10).
DIFFERENTIAL DIAGNOSIS
Full blown Gardner's syndrome should be evident from the clinical observations and radiography. Some entities which can show multiple radio-dense masses or odontomas with impacted and supernumery teeth in the orthopantomograph are cleidocranial dysplasia, osteo-cemental dysplasia and periapical osteo-cemental dysplasia. Different diseases which produce intestinal polyposis include juvenile colonic polyposis, Turcot's syndrome (characterised by the polyps in the colon and rectum and malignant tumours in the central nervous system - gliomas-), Cowden's syndrome (dominant autosomal hereditary multiple hamartomatosis, consisting of trichilemomas which originate in the hair follicle cells) and Peutz-Jeghers syndrome (haemorrhagenic gastrointestinal polyposis and melanin spots on the lips and oral mucosa; autosomal dominant hereditary), among many others. Although the majority of the polyps that appear in Peutz-Jeghers syndrome are localised in the small intestine, some also appear in the large intestine. However, only Gardner's syndrome presents with all, or the majority, of the disturbances referred to in the tetrad of osteomas, fibrous tumours, sebaceous cysts and intestinal polyposis (4,5).
DIAGNOSIS
In establishing a diagnosis, the patient must be examined in great detail. It should be ascertained if there are sebaceous cysts and masses corresponding to fibrous tumours. It is recommended that cranial x-rays and orthopantomographs (panoramic), are performed. If radio-opaque bone lesions or osteomas are seen, it is recommended to obtain a biopsy from at least one of them for further identification. Tests such as gastrointestinal barium meal and opaque enema, to study the distal intestine should be carried out. If there are any suspected findings or the detection of an established Gardner's syndrome, colonoscopy must be performed (4,7). A genetic study of the APC gene is recommended in every individual suspected with FAP or Gardner's syndrome.
TREATMENT
No aetiological treatment for FAP or Gardner's syndrome exists, and only the symptoms can be treated. Given that practically 100% of patients affected by FAP can develop a colorectal cancer, the diagnosis of Gardner's syndrome normally requires a prophylactic colectomy. In some cases serial studies with colonoscopies are carried out, to delay the colectomy, but these studies carry the risk of not detecting a malignant transformation. Although osteomas do not need to be extirpated, they are often removed, due to their appearance and the interference contributed by the mobility. Given the genetic basis of the syndrome, new osteomas can appear after a few months or years. Sebaceous cysts are extirpated at the request of the patient. Fibrous and fibromatous tumours usually divide, but their capacity for local infiltration requires cryotherapy and total extirpation with a margin of safety of 1cm (4).
PROGNOSIS AND FOLLOW UP
In patients with Gardner's syndrome, who are diagnosed early and a colectomy is performed, the prognosis is normally very good. They tend to have an almost normal life. However, follow up should be carried out every 6 – 12 months, patients as well as families, given that the heredity is dominant autosomal in character (4). It has been shown that the maxillofacial radiographic study (orthopantomograph, computerised tomography) can be of great use in the early diagnosis of this pathology and earlier more timely instigation of treatment and follow up of the patients and their families (7,8), although only the concomitant existence of intestinal polyposis allows a definitive diagnosis of the syndrome (10). Once the clinical diagnosis is performed, the genetic study will have the last word, although this also has its limitations (3).
CLINICAL CASE
Female patient, 42 years of age, came to clinic presenting with unspecific pain, in the lower right hemiarcade (fourth quadrant), of several days duration. On clinical examination the pain was confirmed by tapping the first molar -4.6-. The patient said she had suffered several painful episodes over approximately one year, which resolved themselves spontaneously. Palpation of the soft and hard structures of the maxillofacial area showed the presence of protuberances of a bony consistency, compatible with exostosis, of some 5 mm in diameter, in the upper left hemiarcade (second quadrant, at the level of the bottom of the vestibule of the first molar), as well as in the lower left hemiarcade (third quadrant), near the mandibular angle, which corresponded to the previously mentioned description: “ in the form of drop, at the level of the lower border of the gonion”.
On performing a periapical x-ray of the painful zone (molar 4.6), radio-opaque cotton wool like images were seen. One of them extended from the mesial of the root of 4.7 to the space between the radicle apices of the 4.6, approaching the dental canal, until reaching the level of the mesial apex of this molar. The other, positioned nearer the crown, occupied part of the space between the roots of the 4.6 and 4.5 teeth (Figure 1).
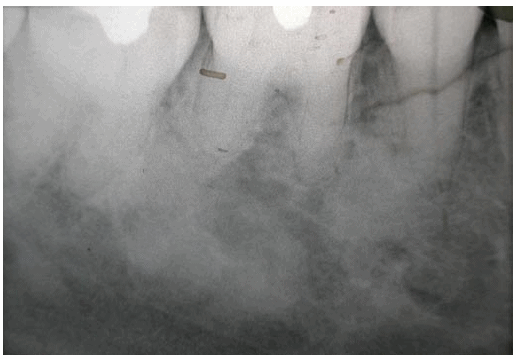
Fig. 1. Periapical x-ray of the painful zone (molar 4.6).
On questioning, the patient told of being affected by familial adenomatous polyposis, a variant of Gardner's syndrome, diagnosed in puberty. She was surgically intervened when 21 years old, total colectomy with ileorectal anastomosis being performed. She also presented with epidermoid cysts on the back and gastric polyps. The genetic study of the patient identified the existence of a mutation in exon 9 of the APC gene which gave rise to a change in the amino acid at the terminal codon (W421X), producing a truncated protein. This genetic disturbance is responsible for the Familial Adenomatous Polyposis described in this family. The genetic study identified that her two children were carriers of the same genetic mutation.
A radiographic study was carried out on the three carrier members of the family (mother and the two children). On the orthopantomograph of the patient, well defined various radio-opaque images of variable sizes and with similar characteristics to those described in the intra-oral periapical projection were seen, and which extended throughout the whole mandibular (Figure 2).
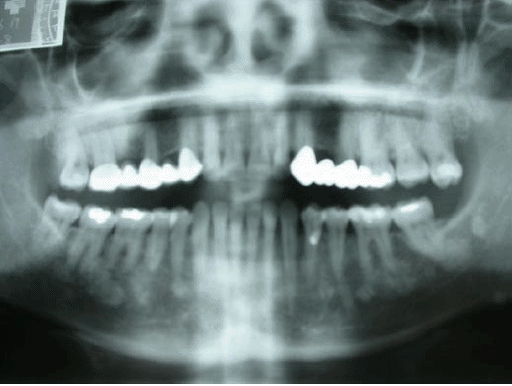
Fig. 2. Orthopantomograph of the patient.
In an orthopantomograph of the daughter, the presence of multiple radio-dense mandibular lesions was confirmed. In the orthopantomograph of the son, subjected to orthodontic treatment, inclusion of the 2.3 canine tooth was observed.
The family history is ample, as shown in the schematic, relating to 1999 (Figure 3). It can be shown, as in this family, despite obvious clinical cases and having identified the mutation responsible for the disease, there are many family members who have not had access to a genetic study to find out whether they are carriers or not of the familial genetic mutation in the family, nor have they had access to the identification of the disease by performing a colonoscopy.
In these families, the extra-colonic phenotype findings, such as the osteomas, can allow the identification of the individuals with a high risk of having inherited the disease and , therefore, increases the need to begin differential diagnostic tests.
DISCUSSION
The importance of the described pathology lies in the great potential for the intestinal polypoid lesions to become malignant. The clinical example put forward tries to illustrate a situation, in which different medical specialities can contribute data and participate in the early diagnosis. This, at least, should allow carrying out preventive treatment or, in the initial phases of the disease, it should contribute to improving the prognosis.
The condensed bone lesions, in the form of osteomas or enostosis, can cause differential diagnosis problems with different nosology entities, such as those which have already been indicated earlier (13). However, the relative ease with which it is clinically or radiologically detected, constitutes a very useful tool for the early diagnosis. Odontologists and stomatologists have much to contribute in this sense, as well as all the specialists in diagnostic imaging techniques. Studies such as those carried out by Thakker et al., in 1995 (7), or Herrmann et al., in 2003 (10), who took into account extra-digestive signs, such as osteomas, deserve a mention as contributions to the early diagnosis of FAP.
Cutaneous and ocular lesions are also easy to examine, so dermatologists, ophthalmologists and other health professionals can likewise contribute to its early diagnosis.
Examination of the digestive system is somewhat more difficult, as the exploratory techniques can appear very troublesome to the patient and it is not always a suitable way to examine, just like in the genetic study. These are, however, the most important investigations for the control of this pathology.
In our hands, as health professionals, it is important, therefore, to know how to adequately direct the patient to those professionals most appropriate to each case.
REFERENCES
1. Online Mendelian Inheritance in Man. OMIM (TM 6 / 7 / 2001). John Hopkins University: Baltimore, MD. MIM Number: *175100: World Wide Web URL: http://www.ncbi.nlm.nih.gow/omim/ [ Links ]
2. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Human Molecular Genetics 2001; 10: 721-33. [ Links ]
3. Bisgaard ML, Ripa R, Knudsen AL, Bulow S. Familial adenomatous polyposis patients without an identified APC germline mutation have a severe phenotype. Gut 2004; 53: 266-7. [ Links ]
4. Marx RE, Stern D. Gardner Syndrome. En: Oral and Maxillofacial Pathology. A Rationale for Diagnosis and Treatment. Chicago: Quintessence Publishing Co, Inc.; 2003. p. 776-8. [ Links ]
5. Matteson SR. Tumores benignos de los maxilares. En: White SC, Pharoah MJ. Radiología oral. Principios e interpretación. Madrid: Elsevier Science; 2002. p. 378-419. [ Links ]
6. Sayan NB, Üçok C, Karasu HA, Günhan Ö. Peripheral Osteoma of the oral and maxillofacial region: a study of 35 new cases. J Oral Maxillofac Surg 2002; 60: 1299-301. [ Links ]
7. Thakker N, Davies R, Horner K, Armstrong J, Clancy T, Guy S et al. The dental phenotype in familial adenomatous polyposis: diagnostic application of a weigheted scoring system for changes on dental panoramic radiographs. J Med Genetics 1995; 32: 458-64. [ Links ]
8. Aggarwal VR, Sloan P, Horner K, Macfarlane TV, Clancy T, Evans G et al. Dento-osseous changes as diagnostic markers in familial adenomatous polyposis families. Oral Diseases 2003; 9: 29-33. [ Links ]
9. Payne M, Anderson JA, Cook J. Gardner's syndrome - a case report. Br Dent J 2002; 193: 383-4. [ Links ]
10. Herrmann SM, Adler YD, Schmidt-Petersen K, Nicaud V, Morrison C, Paul M et al. The concomitant occurrence of multiple epidermal cysts, osteomas and thyroid gland nodules is not diagnostic for Gardner syndrome in the absence of intestinal polyposis: a clinical and genetic report. Br J Dermatol 2003; 149: 877-83. [ Links ]
11. Galle TS, Juel K, Bulow S. Scand. J Gastroenterol 1999; 34: 808-12. [ Links ]
12. Cervantes Ruipérez A: Poliposis adenomatosa familiar. En: Rodés Teixidor J, Guardia Massó J (eds.). Medicina Interna (tomo I). Barcelona: Masson; 1997. p. 1399-1400. [ Links ]
13. White SC, Pharoah MJ. Oral radiology. Principles and interpretation. St Louis, Missouri: Mosby; 2004. p. 410-57. [ Links ]

